










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.22 no.3 Medellín July/Sept. 2009
ARTÍCULO DE REVISIÓN
Psoriasis: revisión del tema con énfasis en la inmunopatogénesis
Psoriasis: a review with emphasis on immunopathogenesis
Carolina Giraldo Sierra1, Margarita María Velásquez Lopera2,3
1Médica, Residente de segundo año de Dermatología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. carolinagiraldos@hotmail.com
2Médica, Dermatóloga, Doctora en Ciencias Biomédicas con énfasis en Inmunología, Universidad de Antioquia, Medellín, Colombia. mmvelasquez@yahoo.com
3Grupo de Investigación Dermatológica, GRID, Sección de Dermatología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. Teléfono (574) 2637667
Resumen
La psoriasis es una de las enfermedades cutáneas más frecuentes pues afecta al 2–3% de la población mundial. Es autoinmune, específica de órgano, crónica y recurrente, desencadenada por factores externos en individuos con predisposición genética. En su inmunopatogénesis se ha descrito una falla en la regulación de la respuesta inmune frente a antígenos aún no bien identificados. Tiene diversas presentaciones clínicas e influye desfavorablemente en la calidad de vida de los pacientes. La comprensión de sus fundamentos inmunopatogénicos ha permitido poner en práctica nuevas estrategias de tratamiento como la terapia biológica. En este artículo se revisan los aspectos fundamentales de la inmunopatogénesis de la psoriasis y sus características epidemiológicas, clínicas e histopatológicas, así como las varias opciones terapéuticas.
Palabras clave
Inmunidad innata, Inmunidad adquirida, Inmunopatogénesis, Psoriasis, Terapia biológica
SUMMARY
Psoriasis is one of the most frequent skin diseases. Worldwide, it affects 2 to 3% of the population. It is an organ–specific, chronic, recurrent, autoimmune disease, triggered by external factors in individuals with a genetic predisposition. In its immunopathogenesis a lack of regulation of the immune response to unidentified antigens has been described. Psoriasis has many different clinical presentations and produces an important decrease in the quality of life. Understanding its immunopathogenetic bases has led to new therapeutic strategies such as the biological approaches. This review includes basic immunopathogenetic aspects of psoriasis, as well as the epidemiological, clinical and histopathological characteristics of the disease. Therapeutic options are also included.
Key words
Adaptive immunity, Biological therapy, Immunopathogenesis, Innate immunity, Psoriasis
INTRODUCCIÓN
La piel es el órgano más extenso del cuerpo humano y cumple importantes funciones como barrera física e inmunológica, para lo cual cuenta con un sistema inmune propio constituido por las células de Langerhans, las células del infiltrado dérmico, los queratinocitos y las células endoteliales, entre otras. Debido a su función de barrera, la piel es el blanco de muchos agresores (infecciosos, climáticos, químicos, térmicos y mecánicos) por lo que, además de la función protectora, debe mantener un delicado equilibrio entre las respuestas inmunes efectora y reguladora para conservar la homeostasis. En la actualidad se acepta que la psoriasis es una enfermedad autoinmune específica de órgano en la cual se han identificado alteraciones de la regulación inmune en individuos con predisposición genética.1,3
HISTORIA
Las primeras descripciones médicas de la psoriasis aparecen en el Corpus Hippocraticum, escrito en los siglos V y IV a. C., en el cual se utilizan indistintamente los términos Psora que significa 'enfermedad que rasca' y Lepra.2,4 Posteriormente se la describe entre los 40 tipos diferentes de 'impétigos' en la Enciclopedia de la Medicina (Celso 25 a. C. a 50 d. C) y como Lepra graecorum y Psora leprosa (Robert William, 1801). En 1841, Ferdinand von Hebra agrupa las lesiones descritas previamente en una misma entidad nosológica denominándola psoriasis.5,6
EPIDEMIOLOGÍA
La psoriasis es una enfermedad de distribución mundial cuya tasa de prevalencia es de 2–3% según el país; es menor hacia el Ecuador y aumenta hacia los polos.7,8 En Europa central su prevalencia es de 1,5%, en América del norte, de 2% y en Sudamérica, de 0,97%.6,7 Dicha tasa también está influida por las características étnicas o raciales: es menor en negros, según lo descrito en un estudio estadounidense.9
La psoriasis afecta tanto a la población adulta como a la infantil; en un estudio de 2.147 pacientes se hallaron dos picos de frecuencia: a los 22 y a los 55 años,10 sin diferencias por sexo.7 En la actualidad se reconocen dos tipos de psoriasis, a saber: el I que comienza antes de los 40 años, ocurre en el 75% de los casos, es más grave y tiene una importante predisposición genética; y el II, menos grave, que comienza después de los 40 años.7,10,11 La psoriasis es una enfermedad crónica, con recaídas y remisiones; el 39% de los pacientes logran la remisión completa del cuadro clínico entre 1 y 54 años después del comienzo de la enfermedad.12
No se han informado estudios que demuestren una relación directa entre la psoriasis y una disminución de la expectativa de vida; sin embargo, en los pacientes con psoriasis se ha descrito mayor incidencia de enfermedades linfoproliferativas, metabólicas, cardíacas y autoinmunes.13
FACTORES DESENCADENANTES
El trauma físico, el estrés, algunos medicamentos, cirugías e infecciones son disparadores del episodio inicial y de las recurrencias de la psoriasis.7
Factores ambientales
La radiación ultravioleta A (UVA) tiene un efecto benéfico en la psoriasis.2,6
Trauma físico
Uno de los signos semiológicos más importantes de la psoriasis es el fenómeno de Koebner, descrito en 1827, que consiste en el desarrollo de lesiones en sitios de microtrauma repetitivo. Dicho fenómeno está relacionado con la liberación de citoquinas proinflamatorias y el desenmascaramiento de autoantígenos.6 Debido a este fenómeno, las placas de psoriasis se presentan con mayor frecuencia en el cuero cabelludo, rodillas y codos.2
Medicamentos
Se han asociado con recurrencias de psoriasis los siguientes medicamentos: beta–bloqueadores, inhibidores de la enzima convertidora de angiotensina (IECA), antimaláricos y litio.2,7 Los mecanismos se conocen solo parcialmente: los beta–bloqueadores inducen hiperproliferación epidérmica asociada a disminución del AMP cíclico intraepidérmico; el litio aumenta la concentración de citoquinas proinflamatorias al reclutar leucocitos, y la cloroquina bloquea la transglutaminasa epidérmica, involucrada en la diferenciación terminal de los queratinocitos.14,15
Infecciones
Se ha encontrado una asociación del 56 al 85% entre la infección respiratoria alta por Streptococcus pyogenes (estreptococo beta hemolítico del grupo A) y la psoriasis aguda en gotas; se ha postulado que las endotoxinas bacterianas pueden actuar como superantígenos dando lugar a una cascada compleja de activación que involucra linfocitos T (LT), macrófagos, células de Langerhans y queratinocitos;2,7,16 además los antígenos estreptocóccicos comparten epítopes con las proteínas de los queratinocitos produciendo reacciones cruzadas que conducen a activación celular.5,16
La infección por el virus de la inmunodeficiencia humana de tipo 1 (VIH–1) puede desencadenar una psoriasis con dos patrones clínicos: localizado, en placas o en gotas extensas, y difuso acompañado de queratodermia palmoplantar. La psoriasis puede ser la primera evidencia de la infección por VIH.2,6
Otros factores
El estrés y el alcohol empeoran la psoriasis hasta en el 40% de los pacientes.2
INMUNOPATOLOGÍA
La psoriasis es una enfermedad autoinmune específica de órgano que ocurre en individuos predispuestos genéticamente. Las causas últimas de este proceso autoinmune, inflamatorio e hiperproliferativo, entre los diferentes modelos inmunopatológicos aceptados en la actualidad, son: una reacción de inmunización e interacciones de la inmunidad innata y la adaptativa que llevan a la producción de diversas citoquinas proinflamatorias, quimioquinas y factores de crecimiento, que generan una respuesta hiperproliferativa de los queratinocitos.3,4,17
Con base en lo anterior se ha descrito un modelo de reacción de inmunización en el que se describen tres fases:5,6
Fase de sensibilización: las células de Langerhans y las células dendríticas (CD) dérmicas captan, procesan y presentan los antígenos para activar los linfocitos T, generando células de memoria central y de memoria efectora. Se desconoce el antígeno que inicia esta respuesta pero se ha propuesto la posible participación de superantígenos como la proteína M del S. pyogenes, la queratina 17 (K17), los patrones moleculares asociados a proteínas de choque térmico (HSP, heat shock proteins) y los glicolípidos. En esta fase no se presentan lesiones.
Fase silenciosa: es de duración variable; en ella no hay lesiones clínicas aparentes ni un fenómeno inmunológico característico.
Fase efectora: se presenta infiltración de la dermis por diferentes tipos de células que luego se activan (monocitos, macrófagos, neutrófilos, células T, células dendríticas), y se generan mecanismos efectores de la inmunidad innata y la adaptativa. La queratina 17 (K17) tiene reactividad cruzada con la proteína M del estreptococo, por lo que la sensibilización podría ocurrir en el curso de una infección estreptocóccica y perpetuarse por reacción a la K17. En esta fase se presentan inflamación, angiogénesis y una respuesta reparativa de los queratinocitos con hiperproliferación y merma de la maduración.
En la fase de sensibilización no está claro si el trastorno de los queratinocitos por microtraumas pudiera ser la primera señal activadora que promueva la reparación y la liberación de citoquinas preformadas como la interleuquina 1 (IL–1) y el factor de necrosis tumoral alfa (TNF–a) que activan las células presentadoras de antígenos (CPA); estas, a su vez, reconocen productos microbianos, superantígenos, patrones moleculares asociados a las proteínas de choque térmico (HSP) liberados por los queratinocitos y glicolípidos, que son ligandos de los receptores tipo Toll (TLR).4,6,17
Luego de la activación, las CPA maduran y migran al ganglio linfático regional para la presentación antigénica a los linfocitos T CD4+ (LT CD4+) vírgenes en el contexto del complejo mayor de histocompatibilidad tipo II (MHC II, Major histocompatibility complex). Para la activación de los linfocitos T se requieren las siguientes señales:3, 6,8,17
Señal 1: reconocimiento del antígeno por los linfocitos T mediante su receptor (TCR, T cell receptor). Las CPA presentan el antígeno en el contexto de las moléculas del MHC.
Señal 2: en ella interviene una serie de moléculas coestimuladoras en la superficie de los LT y las CPA, especialmente CD28:CD80/CD86 y moléculas de adhesión como CD2:LFA–3, LFA–1:ICAM–1.
Señal 3: producción de citoquinas por los LT CD4+ activados, especialmente IL–2, con funciones autocrinas y paracrinas que desencadenan la diferenciación de las células T efectoras y la expansión clonal.
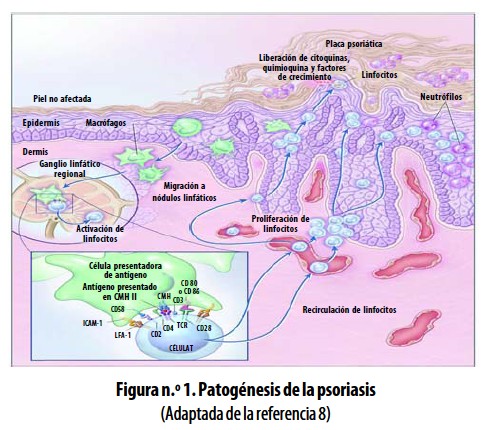
Una vez activados los LT CD4+ se producen expansión clonal y secreción de citoquinas Th1 como interferón gamma (IFN–?), factor de necrosis tumoral ß (TNF–ß, Tumor necrosis factor ß) e IL–2 y se generan células de memoria (CD4+CD45RO+CLA+). En etapas posteriores, los LT CD4+ migran a la piel, son reactivados (posiblemente por autoantígenos) y producen citoquinas, factores de crecimiento y quimioquinas, que tienen diversos efectos sobre los queratinocitos, células endoteliales, neutrófilos, macrófagos, LT CD4+ y CD8+, que determinan la respuesta inflamatoria local y el desarrollo de la enfermedad.4,6,8,17 (Figura n.° 1).
COMPONENTES CELULARES
Células presentadoras de antígenos (CPA)
Hay dos subtipos de CPA en las placas de psoriasis: las células dendríticas (CD) mieloides inmaduras y maduras y, en menor número, las CD plasmocitoides; esto difiere de la piel normal en la que no se encuentran CD plasmocitoides ni CD mieloides maduras.3,18 Las CD se encuentran en la dermis y la epidermis, presentan antígenos y perpetúan la respuesta inflamatoria por producción de citoquinas. Las CD dérmicas son importantes productoras de TNF–a.3,4,18
Polimorfonucleares neutrófilos y macrófagos
En una lesión temprana de psoriasis se observan macrófagos en la epidermis, seguidos de linfocitos y, por último, neutrófilos.2 Estos últimos se localizan en la dermis, pero migran hasta el estrato córneo de la epidermis formando acúmulos, los microabscesos espongiformes de Munro, visibles mediante histopatología;2,8 sus funciones principales incluyen la fagocitosis y la secreción de citoquinas proinflamatorias.
Células NKT
Las células NKT (Natural killer T) expresan TCR con un limitado repertorio (Va24JaQ y Vß11) y receptores de células NK tipo lectina como CD94 y CD161, que les permiten reconocer glicolípidos endógenos presentados por las moléculas CD1. Luego de la activación, las células
Ver (Figura1)
Ver (Figura2)
NKT proliferan y producen IFN–? que favorece el perfil Th1 de citoquinas (IFN–?, IL–2, TNF–a), o IL–4 que favorece el perfil Th2 de las mismas (IL–4, IL–5, IL–13), lo que sugiere su papel en la inmunorregulación.3,19
Se ha informado que en la psoriasis hay aumento de las células NKT circulantes20 y sobreexpresión de CD1d (Cluster of differentiation 1d) en los queratinocitos de las placas psoriáticas en toda la epidermis, a diferencia de la piel sana, en la que lo expresa solamente la capa córnea; la expresión de CD1d depende de IFN–?.18,19,21 Las células NKT se activan al reconocer el complejo CD1d–glicolípido de los queratinocitos y las CPA, produciendo IFN–? y citoquinas Th1 lo que perpetúa la inflamación.3,19
Linfocitos T CD4+
Los LT CD4+ están distribuidos en toda la placa psoriática y principalmente en la dermis papilar. Estas células activadas expresan un fenotipo CD2+, LFA–1, CLA+ y CCR10, que facilita su interacción con moléculas de adhesión del endotelio como son, selectina P, selectina E y CCL27, que permiten su migración y acumulación en la placa psoriática.
Linfocitos T CD8+
Los LT CD8+ que expresan la integrina CD103, cuyo ligando es la cadherina E, migran (exocitosis) hasta la epidermis y permanecen en la placa psoriática;3,7,8 producen IFN–? que es responsable de la persistencia de las lesiones cutáneas.1 A diferencia de la piel en la que predominan los LT CD4+ en una relación CD4+:CD8+ de 2:1, en las articulaciones de los pacientes con artritis psoriática predominan los LT CD8+.22
Queratinocitos
Las placas eritematodescamativas de la psoriasis presentan hiperproliferación de queratinocitos por estímulo de IL–3, IL–6 y factor estimulante de colonias de granulocitos y macrófagos (GM–CSF, Granulocytemacrophage colony–stimulating factor), lo que lleva a que la transición desde la capa basal hasta la capa córnea pase de 28–30 días en individuos sanos, a 3–4 días en pacientes con psoriasis. Lo anterior altera la diferenciación, lo que se refleja en la ausencia de capa granular en la epidermis y paraqueratosis.7,8 Por otro lado, se han demostrado alteraciones en la apoptosis de los queratinocitos, debidas al incremento en el nivel de la proteína antiapoptótica Bcl–x ante el estímulo del IFN–?, lo que aumenta la supervivencia de los queratinocitos.23 También se han informado alteraciones en la expresión y el balance entre las proteínas proapoptóticas como Bax y las antiapoptóticas como Bcl– 2 y Bcl–x, en las que el efecto final es la resistencia a la apoptosis.24
Células reguladoras
La regulación inmune es un mecanismo de tolerancia para prevenir y controlar la autoinmunidad. La ejercen diferentes tipos de células reguladoras; de ellas, las mejor caracterizadas son las naturales CD4+CD25high (Treg) y las adaptativas Tr1 y Th3. El efecto regulador se lleva a cabo por mecanismos dependientes del contacto célulacélula o de factores supresores solubles como la IL–10 y el factor transformante del crecimiento beta (TGF–ß, Transforming growth factor ß).3,25
Sugiyama y colaboradores26 informaron que en la sangre periférica de pacientes con psoriasis es normal el número de células T reguladoras CD4+CD25high, pero que la función reguladora está disminuida. Igualmente describieron un aumento del número de células reguladoras en la placa psoriática, lo que podría sugerir que las alteraciones de la regulación inmune participan en la inmunopatogénesis de la enfermedad.3 Comprender las alteraciones de la regulación inmune en la psoriasis podría en el futuro contribuir al desarrollo de la inmunoterapia.
INMUNIDAD INNATA Y CITOQUINAS
Receptores tipo Toll (TLR)
Los TLR (Toll–like receptors) son una familia de proteínas de membrana que actúan como receptores de reconocimiento de patrones moleculares asociados a patógenos (PAMP, Pathogen–associated molecular patterns) y estimulan la respuesta inmune innata. Hasta ahora se han identificado 10 TLR funcionales en seres humanos.25 Cuando un ligando estimula a su TLR se inicia una cascada de señalización que lleva a movilizar el factor nuclear kappa B (NF–?B) al núcleo celular e induce la transcripción de muchos factores de crecimiento y citoquinas.1,3,27
Se ha sugerido que en la psoriasis los TLR pueden estar involucrados en el reconocimiento de agentes exógenos, productos microbianos o ligandos propios como la fibronectina o patrones moleculares asociados al choque térmico (HSP, heat–shock proteins).3 En un estudio de inmunohistoquímica en que se compararon pieles psoriática y normal, se halló que los TLR–1 y 2 están incrementados en los queratinocitos de la capa basal, mientras que el TLR–5 está disminuido en la piel psoriática.28 En la psoriasis, la expresión de HSP por los queratinocitos induce la activación de las células de Langerhans al unirse al TLR–4, estimulando su maduración y la secreción de TNF–a e IL–12.3,27
Citoquinas
En la psoriasis las citoquinas y las quimioquinas reclutan a los LT y perpetúan la inflamación.8 La placa psoriática se caracteriza por la presencia de citoquinas Th1 como IFN–?, IL–2 y TNF–a, así como de CPA, que ayudan a localizar el infiltrado inflamatorio produciendo IL–18, IL– 23 y TNF–a. A su vez, IL–18 e IL–23 incrementan la producción de IFN– ? por los LT activados.3,29,32
TNF–a: esta citoquina proinflamatoria merece especial atención en la psoriasis debido a que su sobreproducción por diferentes tipos de células como macrófagos, monocitos, neutrófilos, mastocitos, células NK, LT activados, células de Langerhans, células dendríticas dérmicas, queratinocitos y células endoteliales, lleva a la activación de la inmunidad innata y de la adaptativa, que conduce a inflamación crónica, daño tisular y proliferación de los queratinocitos.3,28 El TNF–a induce la activación de las células blanco y la translocación al núcleo del factor de trascripción NF–?B, lo cual a su vez induce la transcripción de TNF–a, GM–CSF, óxido nítrico–sintetasa, ciclo–oxigenasa 2, IL–2 e IL–2R, IL–6, la molécula de adhesión intercelular 1 (ICAM–1, Intercellular adhesion molecule–1), la molécula de adhesión celular vascular 1 (VCAM–1, Vascular cell adhesion molecule–1), la selectina E y la cadena ß del TCR; estos efectos se traducen en incremento de la adhesión, migración y proliferación de linfocitos y, al mismo tiempo, contribuyen a la respuesta hiperproliferativa de los queratinocitos.3,22,31 Con el advenimiento de la terapia biológica anti–TNF–a para la psoriasis, se han estudiado más ampliamente los efectos de esta sustancia en dicha enfermedad.
Interleuquina 17: la IL–17, producida por los LT CD4+ activados, que se denominan células Th17, tiene actividad sobre las células epiteliales. Entre sus funciones se han descrito, dependiendo de su cantidad y concentración, el aumento de IL–6 que induce proliferación de queratinocitos, de IL–8 la cual es quimiotáctica para neutrófilos y de ICAM–1. A diferencia de la piel sana, en biopsias de lesiones psoriáticas se puede detectar ARNm de IL–17, lo que indica que esta interleuquina puede amplificar y mantener la inflamación en la psoriasis.30,31,33
Quimioquinas
Las quimioquinas participan en el reclutamiento de células a la placa psoriática y en la liberación local de citoquinas y factores de crecimiento.4,32 De estas sustancias, las que participan en la inmunopatogénesis de la psoriasis incluyen CCL17, CXCL9 (monoquina inducida por interferón ?), CXCL10, CCL22, CCL5, CCL27 (quimioquina atrayente de la célula T cutánea), CCL20 (proteína inflamatoria del macrófago), CCL19 y CXCL8 (quimioquina atrayente del neutrófilo).4,8,32
Moléculas de adhesión
El primer paso para el reclutamiento de los leucocitos desde la luz vascular hacia el tejido es mediado por una familia de moléculas de adhesión denominadas selectinas; una vez activado, el endotelio expresa selectina E (CD62E) y selectina P (CD62P) que se unen a ligandos en la superficie de los LT como el antígeno común leucocitario asociado a linfocitos T (CLA, Common leukocyte antigen); en un segundo paso, los leucocitos aumentan la firmeza de la adhesión al endotelio por interacciones entre ß2 integrinas (LFA–1, Lymphocyte function–associated antigen–1) y las moléculas de adhesión ICAM–1 (CD54) y VCAM–1.2,10 La expresión de estas moléculas aumenta por las citoquinas proinflamatorias IL–1 y TNF–a producidas durante la psoriasis.2,27
Factores de crecimiento
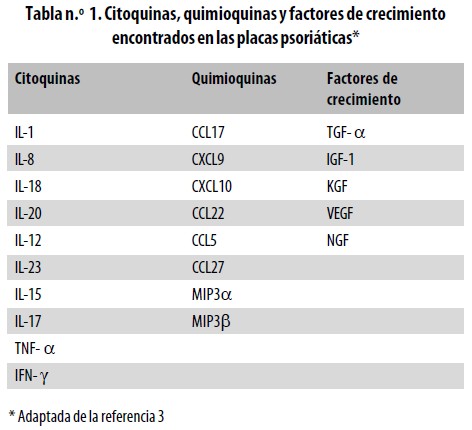
Diversos factores de crecimiento están presentes en las lesiones de psoriasis, entre estos, el factor de crecimiento vascular endotelial (VEGF, Vascular endothelial growth factor), el factor transformante del crecimiento a (TGF– a, Transforming growth factor–a), el factor de crecimiento asociado a insulina–1 (IGF–1, Insulin growth factor), el factor de crecimiento de queratinocitos (KGF, Keratinocyte growth factor), el factor de crecimiento neural (NGF, Neural growth factor) y la IL–20. Estos factores de crecimiento contribuyen a la hiperplasia epidérmica, la resistencia a la apoptosis y la proliferación de linfocitos.3 Otros cambios como la elongación y tortuosidad vasculares se deben a angiogénesis no regulada en respuesta al TGF–ß, al factor de crecimiento derivado de plaquetas (PDGF, Platelet–derived growth factor) y al factor de crecimiento del endotelio vascular (VEGF).19,32 (Tabla n.° 1).
Ver (Tabla1)
BASES GENÉTICAS DE LA PSORIASIS
La búsqueda de las bases genéticas de la psoriasis se fundamentó en los trabajos de Lomholt en 1963, quien exploró la influencia del ambiente y la herencia en residentes de las islas Faroe (Dinamarca), y la afección de muchos familiares. Este autor describió una concordancia del 70% entre gemelos monocigóticos afectados por psoriasis.34
El primero de los locus relacionados con esta enfermedad fue el HLA–Cw6. Los individuos heterocigotos para dicho locus tienen un riesgo relativo (RR) de desarrollar psoriasis de 8,9, comparado con 23,1 en los homocigotos.4 Los alelos HLA–Cw6, HLA–B57 y HLA–DRB1 se han asociado con la psoriasis tipo I y el alelo HLA–Cw2, con la tipo II.7,27
El estudio del genoma humano ha revelado al menos 9 regiones candidatas a contener genes que determinan susceptibilidad para psoriasis (PSORS 1–9); entre ellas, PSORS–1, ubicada en el cromosoma 6 (6p21), es el principal determinante de psoriasis.35 A pesar del estudio de esta región, no se ha identificado el gen implicado; los primeros dos locus candidatos son el HLA–Cw6 ya descrito y el gen que codifica la corneodesmosina, que funciona como una proteína de adhesión de los queratinocitos, importante en su diferenciación terminal en la epidermis.4 La segunda región candidata a contener genes que determinan susceptibilidad para psoriasis es PSORS–2, ubicada en el cromosoma 17 (17q24–q25), cuyos dos genes candidatos son: SLC9A3R1 cuyo disbalance puede deteriorar la sinapsis inmunológica, con lo que se incrementa el tiempo de presentación antigénica y se da lugar a hiperactivación de LT;4,36 el segundo gen identificado es el NAT9, un miembro de la familia N–acetil transferasa, que altera los patrones de glicosilación de varias proteínas inmunorreguladoras, incluyendo los componentes de la sinapsis inmunológica.4,37
Por esta razón los genetistas han enfocado en la sinapsis inmunológica las bases moleculares de la autoinmunidad, y han sugerido que alteraciones en la señalización a través del TCR, que es un elemento importante en la tolerancia central, pueden llevar a la aparición de células T con alta afinidad por autoantígenos; una vez activadas, estas células podrían interactuar con las CPA y los queratinocitos.4,37 En la psoriasis y la artritis psoriática, las células T autorreactivas no destruyen los queratinocitos ni los sinoviocitos, sino que los estimulan a proliferar, lo que podría deberse a una predisposición genética que lleva a la activación incontrolada de dichas células.36
Con base en los análisis de familias con más de dos miembros afectados por psoriasis, se han propuesto otras regiones candidatas a contener genes que determinan susceptibilidad para esta enfermedad; entre ellas, las ubicadas en los cromosomas 4q34 (PSORS–3), 1q21 (PSORS–4), 3q21 (PSORS–5), 19p13 (PSORS–6), 1p (PSORS– 7) y 4q31 (PSORS–9).4,36,37
Varios de los locus implicados son compartidos por otras enfermedades inflamatorias y autoinmunes, como diabetes mellitus tipo I, esclerosis múltiple, dermatitis atópica, enfermedad inflamatoria intestinal, asma y artritis reumatoide, lo que sugiere que hay mecanismos genéticos comunes en dichas enfermedades.35
PSORIASIS Y SINDROME METABÓLICO
En años recientes se ha descrito que la psoriasis está asociada a factores de riesgo para el síndrome metabólico, como obesidad, diabetes mellitus tipo 2 (DM2), dislipidemia e hipertensión; se ha propuesto que los factores genéticos comunes entre obesidad y psoriasis, asociados a un estilo de vida no saludable, llevan a sobrepeso e inducen cambios en la producción y el metabolismo de citoquinas proinflamatorias como IL–6 y TNF–a. Estas citoquinas, además de contribuir a la inmunopatogénesis de la psoriasis, están relacionadas con la resistencia a la insulina que genera intolerancia a la glucosa y DM2, e intervienen en el metabolismo de los lípidos induciendo un perfil aterogénico con incremento del colesterol total, los triglicéridos y las lipoproteínas de baja densidad (LDL) y disminución de las de alta densidad (HDL). De otro lado, la psoriasis está asociada a hipertensión arterial debido al incremento de la enzima convertidora de angiotensina, la endotelina 1 y la renina, lo que altera la relación angiotensina/bradiquinina e induce constricción arterial. Teniendo en cuenta lo anterior, es importante comprender que los pacientes con psoriasis requieren un enfoque multidisciplinario para el tratamiento de las manifestaciones cutáneas, articulares, metabólicas y cardiovasculares.38
PRESENTACIÓN CLÍNICA
La psoriasis es una enfermedad eritematodescamativa con morfología, distribución, gravedad y curso variables. Las lesiones que la caracterizan son placas bien delimitadas de borde nítido y distribución simétrica, con escamas plateadas no cohesivas, sobre una base eritematosa, que producen prurito y sensación de quemadura y en las que se encuentran el signo de Auspitz (cuando se quitan las escamas hiperqueratósicas aparecen pequeñas gotas de sangre sobre la superficie de la lesión) y el fenómeno de Koebner (desarrollo de lesiones en sitios de microtrauma repetitivo).2,14
La extensión de la psoriasis se calcula según el índice PASI (Psoriasis Area Severity Index), que relaciona la extensión, el eritema y el grosor de las escamas presentes en el área afectada; el puntaje máximo es de 75 y según su valor la enfermedad se clasifica en leve (menos de 10), moderada (10–50) y grave (más de 50).10
Tipos clínicos
Psoriasis en placas o vulgar: es la forma más frecuente de la enfermedad; se caracteriza por placas eritematosas bien delimitadas, cubiertas de escamas plateadas, que afectan principalmente las zonas de presión.10,14
Psoriasis eruptiva (en gotas): representa el 2% de los casos; es autolimitada y se manifiesta con placas pequeñas (0,5–1,5 cm de diámetro); es más frecuente en niños y adolescentes luego de infecciones estreptocóccicas de la vía aérea superior.2,14,39
Psoriasis inversa o flexural: consiste en placas rojas bien delimitadas, sin escamas, que afectan los pliegues, principalmente el inframamario, el perineal y el axilar.14
Eritrodermia psoriática: representa una forma generalizada de la enfermedad, con eritema, edema y menos descamación. Este cuadro clínico lo precipitan infecciones y fármacos; puede producir alteraciones en la termorregulación que llevan a hipotermia, descompensación cardíaca y cambios metabólicos que incluyen hipoalbuminemia, anemia por pérdida de hierro, vitamina B12 y folatos.2,10,14
Psoriasis pustular generalizada (de von Zumbush): es una forma menos frecuente de la enfermedad en la que se observan múltiples pústulas de 2–3 mm sobre una base eritematosa, en el tronco y las extremidades, incluidos los lechos ungueales, las palmas y las plantas; puede llegar a ser generalizada con fiebre, dolor y malestar general. La desencadenan infecciones o tratamientos con esteroides tópicos o sistémicos.10,14
Psoriasis palmoplantar o localizada: se presentan pústulas estériles palmoplantares sobre una base eritematosa; es nueve veces más frecuente en mujeres que en hombres y la edad promedio de aparición es entre los 40–60 años. Se ha asociado en el 25% de los casos con la psoriasis vulgar y en el 95% con pacientes fumadores.14
Psoriasis ungueal: se presenta en 25–30% de los pacientes con psoriasis e incluso puede ser un hallazgo aislado; afecta con mayor frecuencia las uñas de las manos que las de los pies. Los cambios consisten en depresiones puntiformes (pits) en la lámina ungueal; máculas amarillentas (manchas de aceite) en la porción distal de la uña, onicolisis y onicodistrofia por compromiso de la matriz ungueal.2,10,14
Psoriasis de las mucosas: infrecuentemente pueden estar afectadas la mucosa del glande y la oral en forma de placas eritematosas, bien delimitadas sin escamas.10,14
TRATAMIENTO
Aunque el objetivo de esta revisión no es presentar una guía de tratamiento, se describen someramente las terapias utilizadas.
Terapia tópica
Antralina: tiene actividad antiproliferativa sobre los queratinocitos, efecto antiinflamatorio sobre la placa psoriática e inhibición de las funciones oxidativas de los neutrófilos.2,8,40
Vitamina D3 y sus análogos: inhiben la proliferación de los queratinocitos, inducen su diferenciación terminal, tienen propiedades antiinflamatorias al inhibir el factor nuclear ?B (NF–?B) y reducen la presentación antigénica porque inhiben la maduración de las CPA.2,8,40
Glucocorticoides tópicos: tienen actividades antiinflamatoria, antimitótica, inmunomoduladora y vasoconstrictora.8,10
Terapia sistémica
Metotrexate (MTX): inhibe la enzima dihidrofolato reductasa, lo que frena la síntesis de purinas y de ADN,2,8,41,42 con merma de la proliferación de linfocitos y queratinocitos, disminución de la secreción de citoquinas como IL–1, IFN–? y TNF–a 43 e inhibición de la activación de los macrófagos.42
Ciclosporina: disminuye la transcripción de citoquinas (IL–2 e IFN ?), quimioquinas y factores de crecimiento. Inhibe la presentación antigénica y la función de los mastocitos (degranulación y producción de citoquinas).2,4,8,29
Retinoides: inhiben la trascripción génica de los queratinocitos disminuyendo así su proliferación y diferenciación.2,42
Mofetil micofenolato: inhibe la inosina monofosfato dehidrogenasa, enzima clave en la síntesis de novo de las purinas. También inhibe la proliferación de los linfocitos T y retrasa su reclutamiento, al disminuir el número de moléculas de adhesión producidas en respuesta al estímulo antigénico.29,39,41
Glucocorticoides sistémicos: tienen efecto inhibidor sobre la transcripción genética de citoquinas (IL–1, IL–2, IL–6, IL–8, IFN–?, TNF–a y moléculas de adhesión), reducen la síntesis de leucotrienos, prostaglandinas y óxido nítrico.41
Leflunomida: tiene propiedades antivirales, antitumorales e inmunosupresoras.44
Fotoquimioterapia (PUVA)
Combina un fotosensibilizante potente como es el 8– metoxipsoraleno (8–MOP) con la exposición a radiación UV (UVA: 320–400 nm); esto suprime irreversiblemente la síntesis de ADN y la mitosis en las células epiteliales, con la subsecuente inhibición de su proliferación, merma de la producción de IL–2, inhibición de la actividad quimiotáctica y merma de la presentación antigénica.2,45 Además, favorece el desarrollo de repuestas Th2 (desviación inmune).43
Terapia biológica
Para el control de los eventos inmunopatológicos de la psoriasis se han identificado, entre otros, los siguientes blancos terapéuticos:8,9,46 interrupir la migración de los LT activados desde el endotelio hasta la dermis y la epidermis; inhibir la activación y proliferación de linfocitos T; modular la liberación y los efectos biológicos de las citoquinas inflamatorias (TNF–a) y promover mecanismos de desviación inmune.
Alefacept: es una proteína de fusión recombinante, que bloquea la interacción LFA–3/CD2 (Lymphocyte functionassociated antigen–3/Cluster of differentiation 2) por una inhibición competitiva, induciendo apoptosis y depleción de las células T de memoria.8,10,43,44,47,48 Estudios multicéntricos, doble ciego, controlados con placebo han demostrado su eficacia en pacientes con psoriasis de moderada a grave.49,50
Efalizumab: es un anticuerpo monoclonal IgG1 humanizado dirigido contra la subunidad CD11a del LFA– 1, que perturba la presentación antigénica y la activación y proliferación celulares.8,9,42,43,47,48,51 Lebwohl y colaboradores demostraron su eficacia al estudiar 597 pacientes con psoriasis de moderada a grave; encontraron una reducción de 75% del índice PASI.52
HuMax–CD4: es un anticuerpo humano contra la molécula CD4 de los LT, que bloquea la interacción con el MHC II. Con este tratamiento, Skov y colaboradores demostraron reducción del PASI y disminución dependiente de la dosis del número de LT CD4+.53
Etarnecept: es una proteína recombinante que inhibe el TNF–a soluble disminuyendo así sus funciones biológicas proiinflamatorias.9,47,48,51 Leonardi y colaboradores estudiaron a 672 pacientes con psoriasis de moderada a grave, y encontraron mejoría del 75% con base en el índice PASI.54
Infliximab: es un anticuerpo monoclonal quimérico que inhibe la actividad del TNF–a al unirse tanto a la forma soluble como a la que está en la membrana celular. Al unirse a esta última activa una vía de señalización intracelular que induce apoptosis. 9,47,48,51
Adalumimab: es un anticuerpo monoclonal de tipo IgG1 humano, dirigido contra el TNF–a; bloquea su interacción con los receptores de la superficie celular p55 y p75, inhibiendo sus funciones biológicas.9,47,48,51
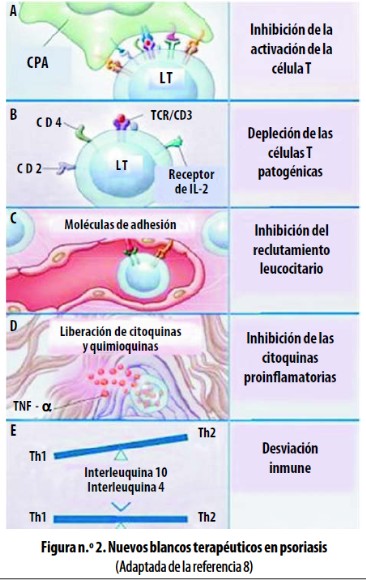
En la figura n.° 2 se resumen los nuevos blancos terapéuticos en la psoriasis.
CONCLUSIÓN
La psoriasis es una enfermedad autoinmune específica de órgano, de carácter crónico, recurrente y complejo; en su aparición y exacerbación participan muchos factores, entre los que se incluyen la predisposición genética, dada por las regiones candidatas PSORS1–9, el clima, las infecciones, los medicamentos, el trauma físico y el estrés. Se induce una falta de regulación en el sistema inmune, desencadenada por antígenos aún no determinados, que lleva a activación y presentación antigénica incontrolada por parte de las CPA a los LT, los cuales secretan citoquinas, principalmente Th1, que reclutan muchas células diferentes y perpetúan la inflamación, pero que principalmente estimulan la proliferación y un cambio en la maduración de los queratinocitos lo que da lugar a la placa psoriática; con base en la comprensión de este proceso, se cuenta con la terapia tópica y sistémica y se ha introducido la terapia biológica que incluye medicamentos bloqueadores de la sinapsis inmunológica y que por ende disminuyen la activación de los LT, como el alefacept, efalizumab y HuMax CD4+; también se dispone de medicamentos que bloquean o cambian el microambiente de citoquinas, especialmente del TNF–a como infliximab, etanercept y adalumimab. El éxito de la terapia biológica anti–TNF–a, además de ayudar al control de la enfermedad, ha contribuido a incrementar el conocimiento sobre la inmunopatogénesis de la psoriasis.
REFERENCIAS BIBLIOGRÁFICAS
1. Robert C, Kupper TS. Inflammatory skin diseases, T cells, and immune surveillance. N Engl J Med 1999; 341: 1817– 1828. [ Links ]
2. Grodjonsson J, Elder J. Psoriasis. En: Wolff K, Goldsmith LA, Katz SI, Gilchrist BA, Paller AS, Lefell DJ, eds. Fitzpatrick's Dermatology in General Medicine, 7ª ed. New York: McGraw–Hill; 2008. p. 169–193. [ Links ]
3. Gaspari AA. Innate and adaptive immunity and the pathophysiology of psoriasis. J Am Acad Dermatol 2006; 54: S67–S80. [ Links ]
4. Nickoloff BJ, Frank O. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J Clin Invest 2004; 113: 1664– 1675. [ Links ]
5. Sabat R. Immunopathogenesis of psoriasis. Exp Dermatol 2007; 16: 779–798. [ Links ]
6. Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature 2007; 445: 866–873. [ Links ]
7. Prinz JC. Psoriasis. En: Bos JD, ed. Skin immune system, Cutaneous Immunology and Clinical Inmunodermatology. 3ª ed. Amsterdam: CRC Press; 2005. p 615–630. [ Links ]
8. Sch$ouml;n MP, Boehncke WH. Psoriasis. N Engl J Med 2005; 352: 1899–1912. [ Links ]
9. Kormeili T, Lowe NJ, Yamauchi PS. Psoriasis: immunopathogenesis and evolving immunomodulators and systemic therapies; U.S. experiences. Br J Dermatol 2004; 151: 3–15. [ Links ]
10. Alegre de Miguel V. Psoriasis. 2004 [consultado agosto 30 de 2007]. Disponible en http://www.uv.es/derma. [ Links ]
11. Christophers E. Psoriasis–epidemiology and clinical spectrum. Clin Exp Dermatol 2001; 26: 314–320. [ Links ]
12. Henseler T, Christophers E. Psoriasis of early and late onset: characterization of two types of psoriasis vulgaris. J Am Acad Dermatol 1985; 13: 450–466. [ Links ]
13. Melski JW, Stern RS. The separation of susceptibility to psoriasis from age at onset. J Invest Dermatol 1981; 77: 474–477. [ Links ]
14. Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis 2005; 64 (Suppl 2): 18–23. [ Links ]
15. Tsankov N, Angelova I, Kazandjieva J. Drug–induced psoriasis. Recognition and management. Am J Clin Dermatol 2000; 1: 159–165. [ Links ]
16. Gudjonsson JE, Thorarinsson AM, Sigurgeirsson B, Kristinsson KG, Valdimarsson H. Streptococcal throat infections and exacerbation of chronic plaque psoriasis: a prospective study. Br J Dermatol 2003; 149: 530–534. [ Links ]
17. Nickoloff BJ, Qin JZ, Nestle FO. Immunopathogenesis of psoriasis. Clin Rev Allergy Immunol 2007; 33: 45–56. [ Links ]
18. Jariwala SP. The role of dendritic cells in the immunopathogenesis of psoriasis. Arch Dermatol Res 2007; 299: 359–366. [ Links ]
19. Watts C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat Immunol 2004; 5: 685–692. [ Links ]
20. Cameron AL, Kirby B, Griffiths CE. Circulating natural killer cells in psoriasis. Br J Dermatol 2003; 149: 160–164. [ Links ]
21. Bonish B, Jullien D, Dutronc Y, Huang BB, Modlin R, Spada FM, et al. Overexpression of CD1d by keratinocytes in psoriasis and CD1d–dependent IFNgamma production by NK–T cells. J Immunol 2000; 165: 4076–4085. [ Links ]
22. DJ Veale, Ritchlin C, Fitzgerald O. Immunopathology of psoriasis and psoriatic arthritis. Ann Rheum Dis 2005; 64: 26–29. [ Links ]
23. Chaturvedi V, Qin JZ, Denning MF. Apoptosis in proliferating, senescent, and immortalized keratinocytes. J Biol Chem 1999; 274: 23358–23367. [ Links ]
24. Koçak M, Bozdogan O, Erkek E, Atasoy P, Birol A. Examination of Bcl–2, Bcl–X and bax protein expression in psoriasis. Int J Dermatol 2003; 42: 789–793. [ Links ]
25. Abbas A, Lichtman A. Inmunología Celular y Molecular. 5a ed. Madrid: Elsevier; 2004. p 216–225 [ Links ]
26. Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, et al. Dysfunctional blood and target tissue CD4CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol 2005; 174: 164–173. [ Links ]
27. Bos JD. Psoriasis, innate immunity, and gene pools. J Am Acad Dermatol 2007; 56: 468–471. [ Links ]
28. Baker BS, Ovigne JM, Powles AV, Corcoran S, Fry L. Normal keratinocytes express Toll–like receptors (TLRs) 1, 2 and 5: modulation of TLR expression in chronic plaque psoriasis. Br J Dermatol 2003; 148: 670–679. [ Links ]
29. Breathnach SM. The skin immune system and psoriasis. Clin Exp Immunol 1993; 91: 343–345. [ Links ]
30. Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin–17 and interferon–gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol 1998; 111: 645–649. [ Links ]
31. McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL–23–IL–17 immune pathway. Trends Immunol 2006; 27: 17–23. [ Links ]
32. Nickoloff BJ, Xin H, Nestle FO, Qin JZ. The cytokine and chemokine network in psoriasis. Clin Dermatol 2007; 25: 568–573. [ Links ]
33. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006; 24: 677–688. [ Links ]
34. Lomholt G. Psoriasis: prevalence, spontaneous course and genetics; a census study on the prevalence of skin diseases on the Faroe islands. Copenhagen: GEC–Gad; 1963, p. 295. [ Links ]
35. Smith CH, Barker JN. Psoriasis and its management. BMJ 2006; 333: 380–384. [ Links ]
36. Krueger JG, Bowcock A. Psoriasis pathophysiology: current concepts of pathogenesis. Ann Rheum Dis 2005; 64 (Suppl 2): 30–36. [ Links ]
37. Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nature Rev Immunol 2005; 5: 699–711. [ Links ]
38. Gottlieb AB, Dann F, Menter A. Psoriasis and the metabolic syndrome. J Drugs Dermatol 2008; 7: 563–572. [ Links ]
39. Martin BA, Chalmers RJ, Telfer NR. How great is the risk of further psoriasis following a single episode of acute guttate psoriasis? Arch Dermatol 1996; 132: 717–718. [ Links ]
40. Afifi T, de Gannes G, Huang C, Zhou Y, Topical therapies for psoriasis: Evidence–based review. Can Fam Physician 2005; 51: 519–525. [ Links ]
41. Nelson RP, Ballow M. Immunomodulation and immunotherapy: Drugs, cytokines, cytokine receptors, and antibodies. J Allergy Clin Immunol 2003; 111: S720– S732. [ Links ]
42. Philipp S, Wolk K, Kreutzer S, Wallance E, Ludwing N, Roewert J, et al. The evaluation of psoriasis therapy with biologics leads to a revision of the current view of the pathogenesis of this disorder. Expert Opin Ther Targets 2006; 10: 817–831. [ Links ]
43. Joshi R. Immunopathogenesis of psoriasis. Indian J Dermatol Venereol Leprol 2004; 70: 10–12. [ Links ]
44. Thappa DV, Laxmisha C. Immunomodulators in the treatment of psoriasis. Indian J. Dermatol Venereol Leprol 2004; 70: 1–9. [ Links ]
45. De Rie MA, Bos JD. Photo (chemo) therapeutic modulation of the skin immune system. En: Bos JD, ed. Skin Immune System, Cutaneos Immunology and Clinical Inmunodermatology. 3ª ed. Amsterdam: CRC Press; 2005. p 771–790. [ Links ]
46. Krueger JG. The immunologic basis for the treatment of psoriasis with new biologic agents. J Am Acad Dermatol 2002; 46: 1–23. [ Links ]
47. Winterfield LS, Menter A, Gordon K, Gottlieb A. Psoriasis treatment: current and emerging directed therapies. Ann Rheum Dis 2005; 64 (Suppl. 2I): 87–90. [ Links ]
48. Weinberg JM, Bottino CJ, Lindholm J, Buchholz R. Biologic therapy for psoriasis: an update on the tumor necrosis factor inhibitors infliximab, etanercept, and adalimumab, and the T–cell–targeted therapies efalizumab and alefacept. J Drugs Dermatol 2005; 4: 544–555. [ Links ]
49. Ellis CN, Krueger GG. Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. N Engl J Med 2001; 345: 248–255. [ Links ]
50. Krueger GG, Papp KA, Stough DB. A randomized, double blind, placebo–controlled phase III study evaluating efficacy and tolerability of two courses of alefacept in patients with chronic plaque psoriasis. J Am Acad Dermatol 2002; 47: 821–833. [ Links ]
51. Dogra A, Sachdeva S. Biologic therapy en psoriasis. Indian J Dermatol Venereol Leprol 2006; 72: 256–265. [ Links ]
52. Lebwohl M, Tyring SK, Hamilton TK, Toth D, Glazer S, Tawfik NH, et al. A novel targeted T–cell modulator, efalizumab, for plaque psoriasis N Engl J Med 2003; 349: 2004–2013. [ Links ]
53. Svok L, Kragballek Z. Humax–CD4: a fully human monoclonal anti CD4 antibody for the treatment of psoriasis vulgaris. Arch Dermatol 2003; 139: 1433–1439. [ Links ]
54. Leonardi CL, Powers JL, Matheson RT, Goffe BS, Zitnik R, Wang A, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349: 2014–2022. [ Links ]
Recibido: octubre 08 de 2008
Aceptado: enero 26 de 2009

