










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.22 no.4 Medellín Oct./Dec. 2009
ARTÍCULO DE REVISIÓN
Epigenética en asma
Epigenetics in asthma
Candelaria Vergara Rivera1,Jorge Mario Sánchez Caraballo2,Beatriz Martínez Alfaro3,Luis Caraballo Gracia4
1MD, Investigadora Asociada, Johns Hopkins University, Division of Allergy and Clinical Immunology, Baltimore, MD, Estados Unidos. candevrivera@yahoo.com.ar
2MD, Estudiante de Maestría en Inmunología, Instituto de Investigaciones Inmunológicas, Universidad de Cartagena, Cartagena, Colombia. jotamsc@yahoo.com
3MSc, Profesor Asociado, Investigador del Instituto de Investigaciones Inmunológicas, Universidad de Cartagena, Cartagena, Colombia. beatri23@yahoo.com
4MD, Director y Profesor Titular, Investigador del Instituto de Investigaciones Inmunológicas, Universidad de Cartagena, Cartagena, Colombia. lucaraballo@yahoo.com
RESUMEN
El asma es una enfermedad respiratoria crónica con alta heredabilidad. Se ha propuesto que en su patogénesis participan varios genes con efectos variables al igual que factores ambientales, y se ha sugerido que los mecanismos epigenéticos pueden mediar parte del efecto de los factores ambientales en el comienzo y la evolución de la enfermedad. La epigenética describe los cambios en la expresión génica heredables durante las mitosis y meiosis que no son codificados en la secuencia de ADN. Ellos incluyen la metilación o desmetilación del ADN y la acetilación, desacetilación, ubiquitinación, sumoilación y fosforilación de histonas, cambios en los microARN y alteraciones cromatínicas. En esta revisión se describen hallazgos que establecen una relación entre algunos mecanismos epigenéticos y el proceso inflamatorio y la exposición a factores ambientales en el asma. Ellos incluyen: el aumento en la actividad de las acetilasas de histonas y de la expresión de las enzimas acetiladoras; la disminución de las enzimas desacetiladoras en los pulmones de individuos asmáticos; el aumento de la expresión del factor nuclear NF–?B durante el proceso inflamatorio alérgico; cambios en la metilación/desmetilación del ADN durante la diferenciación de los linfocitos y la estimulación/supresión de genes como los de la IL–4 y el IFN–?, respectivamente. El humo del cigarrillo, las infecciones bacterianas y virales, la dieta materna y la polución ambiental son otros factores que desencadenan procesos epigenéticos como la acetilación de histonas, la inducción de citoquinas inflamatorias, la inactivación de las desacetilasas de histonas, la polarización de la respuesta inmune hacia el tipo Th2 y una mayor producción de IgE y citoquinas de este perfil. Se revisan también los efectos epigenéticos resultantes de la terapia con corticoides y teofilina y otros factores que podrían influir en el riesgo de asma en la infancia como la ingesta materna de frutas, legumbres, aceites de pescado, vitaminas, minerales y probióticos y el uso de antibióticos durante el embarazo.
Palabras clave
Alergia, Asma, Cromatina, Histonas, Metilación
SUMMARY
Epigenetics in asthma
Asthma is a chronic respiratory disease with a high heritability. It has been postulated that several genes with variable effects are involved in its pathogenesis along with environmental factors. It has been suggested that epigenetic mechanisms can mediate the effects of environmental factors on the onset and progression of the disease. Epigenetics describes inheritable changes in gene expression inherited during meiosis and mitosis that are not encoded in the DNA sequence. They include DNA methylation/ demethylation, acetylation, deacetylation, ubiquitination, SUMOylation and phosphorylation of histones, changes in microARN and alterations of chromatine. In this article we review some findings that establish a relationship between some epigenetic mechanisms and the inflammatory process in asthma and exposure to environmental factors. They include increasing the activity of histone acetyl–transferases and the expression of histone acetylating enzymes, decrease of deacetylating enzymes in the lungs of asthmatics; increased expression of the transcription factor NF–?B in the allergic inflammatory process, changes in methylation/demethlylation of DNA during the differentiation of lymphocytes and the stimulation/ suppression of IL–4 and IFN? genes, respectively. Smoking, bacterial and viral infections, maternal diet and environmental pollution are also factors that trigger epigenetic processes such as histone acetylation and induction of inflammatory cytokines, inactivation of histone deacetytransferases, polarization of the immune response toward the Th2 type and increased production of IgE and cytokines of this profile. We review the epigenetic effects resulting from therapy with corticosteroids and theophylline, and other factors that might influence the risk of asthma in childhood such as maternal intake of fruits, vegetables, fish oils, vitamins, minerals, and the use of probiotics and antibiotics during pregnancy.
Key words
Allergies, Asthma, Chromatine, Histones, Methylation
INTRODUCCIÓN
El asma es una enfermedad respiratoria crónica que se asocia en una alta proporción con aumento del nivel de IgE en la sangre, dirigida contra antígenos ambientales principalmente aeroantígenos. De una manera simplificada, la fisiopatología del asma bronquial comprende una respuesta inmune predominante del tipo Th2 con aumento de las interleuquinas IL–4, IL–5 e IL–13 y disminución del IFN–? y de otras citoquinas del tipo Th1, con participación de eosinófilos, mastocitos y, entre otras células, Th17 reguladoras e iNKTC.1 Ante la exposición al antígeno ambiental, en este caso denominado alergeno, se desarrollan en el individuo genéticamente predispuesto una respuesta mediada por las citoquinas mencionadas y una diferenciación de los linfocitos B hacia células plasmáticas productoras de IgE específica contra el alergeno iniciador y ocurre la denominada sensibilización en la cual la IgE se une a sus receptores sobre los mastocitos;2 en una exposición subsiguiente al mismo alergeno, se genera en el pulmón una respuesta inflamatoria de tipo alérgico que se potencia por las exposiciones posteriores y se perpetúa por la participación de otros mecanismos inflamatorios y de cambios de la estructura normal del tejido pulmonar, fenómeno conocido como remodelación bronquial. Clínicamente, el asma se caracteriza por crisis agudas y períodos asintomáticos en un espectro que varía desde pacientes con asma grave y perenne incompatible con la actividad física hasta algunos casos que presentan una enfermedad intermitente con crisis leves ocasionales y largos períodos libres de síntomas. La edad de inicio y la duración son variables: en algunos casos hay remisión espontánea y definitiva de los síntomas, y en otros, reaparición de la enfermedad en etapas posteriores. Aunque se ha avanzado mucho en las investigaciones de las enfermedades complejas como el asma, se desconocen los mecanismos exactos por los cuales un individuo desarrolla esta enfermedad; sin embargo, a la luz de los conocimientos actuales, se acepta que ella es el resultado de la interacción de un conjunto de factores genéticos, epigenéticos y ambientales. Aspectos más concretos son motivo de intensa investigación, como los determinantes de la edad de comienzo de la enfermedad, los efectos variables de los genes dependiendo del origen paterno del cual provienen, las fluctuaciones en la historia natural de la enfermedad y la discordancia fenotípica de hasta un 25%observada entre gemelos monocigóticos.
Los genes asociados actualmente con el asma son numerosos y tienen múltiples funciones: pueden ser receptores o mediadores de la respuesta inmune o estar relacionados con las estructuras bronquial y vascular. Entre ellos se han identificado como participantes de importancia ADAM33, NPSR1, PHF11, HLA–G mediante estudios de ligamiento genético y mapeo fino.3,5 Por otra parte, los esfuerzos para identificar los factores ambientales relacionados con el asma han dado buenos frutos y es así como se han definido claramente los efectos desfavorables de la polución ambiental, el humo del cigarrillo, los aeroalergenos y las infecciones virales sobre la aparición de asma y en sus exacerbaciones. Sin embargo, otros agentes como la exposición a endotoxinas y las infecciones bacterianas tienen efectos variables y en algunos casos aparentemente contradictorios, dependiendo del momento en que el individuo se exponga y de la dosis; en el caso de las parasitosis intestinales, las opiniones son divergentes. Las investigaciones actuales se orientan a identificar los genes específicos implicados en cada población, cómo se regulan su expresión e interacciones además de la disección de los mecanismos moleculares que median el efecto de los factores ambientales. La combinación de estas estrategias podría finalmente identificar individuos en riesgo y fundamentar la implementación de medidas de control ambiental en la población.
EPIGENÉTICA
Epigenética es el término usado para describir los cambios en la expresión génica heredables durante las mitosis y meiosis que no son codificados en la secuencia del ADN6 sino que se deben a modificaciones postraduccionales del ADN y de las histonas.7 La regulación epigenética es crítica para generar la diversidad de tipos celulares durante el desarrollo y para mantener la estabilidad e integridad de los perfiles de expresión de los diferentes tipos celulares.7Los mecanismos epigenéticos tienen la propiedad de ser autorregulados y en muchos casos reversibles lo que permite cierta plasticidad en la expresión genética dependiendo de los factores externos. Ellos incluyen metilación o desmetilación del ADN y acetilación, desacetilación, ubiquitinación, sumoilación y fosforilación de histonas, cambios en los microARN y alteraciones cromatínicas.8
La metilación y la acetilación de las histonas ocurren en el nucleosoma que es la unidad básica de empaquetamiento de la eucromatina; a partir de él se da la compactación del ADN por varios mecanismos hasta conformar los cromosomas. El nucleosoma está compuesto por un núcleo proteico central conformado por la agrupación de ocho moléculas correspondientes a dos pares de histonas H2A, H2B, H3 y H4 sobre el cual se enrolla un fragmento de ADN de doble hélice de 146 pares de bases que descansa en una unidad de histona H1 localizada fuera del octámero. Los extremos amino de las histonas protruyen en los lados del nucleosoma y son ricos en lisinas (K) y argininas (R); dichos extremos son susceptibles de ser acetilados, metilados y fosforilados. La acetilación la llevan a cabo principalmente las enzimas acetil–transferasas de histonas (HAT, por histone acetyl transferases) y se asocia con una mayor actividad transcripcional. En particular, la acetilación de residuos K de H3 y H4 actúa como una marca molecular para el reclutamiento de enzimas remodeladoras de la cromatina como Brg1, la cual permite el desenrollamiento local de la cromatina y la llegada de otros factores de transcripción, el complejo transcripcional basal y la ARN polimerasa II.7 Por otra parte, la adición de grupos metilo a las histonas permite la adherencia de cromodominios que facilitan el acoplamiento de represores transcripcionales con un efecto opuesto sobre la transcripción comparado con el de la acetilación. En un principio se propuso que el efecto inductor de la acetilación se debía a cambios en las propiedades electrostáticas de las histonas dado que los residuos acetilo aumentan su carga negativa y las hacen menos afines por el ADN, pero en la actualidad se acepta que dicho efecto se debe principalmente a que las modificaciones facilitan el reclutamiento de complejos ARN polimerasas II y bromodominios, los cuales funcionan como adaptadores de factores de transcripción.7 Una propuesta alternativa es la denominada hipótesis del código de histonas según la cual diferentes combinaciones de modificaciones en las histonas pueden resultar en distintos efectos en términos de funciones reguladas por la cromatina.9 Los fenómenos de metilación y acetilación de las histonas son reversibles aunque se acepta que la primera es un cambio más estable que la segunda. La desacetilación la llevan a cabo las desacetilasas de histonas (HDAC, por histone deacetylases) de las cuales se han descrito cuatro clases (I a IV) que tienen distinta distribución celular y especificidad para los varios patrones de acetilación y que funcionan regulando diferentes genes mediante la interacción con diversas proteínas como el correpresor del receptor nuclear (NCoR, por Nuclear receptor corepressor) y el mediador del silenciamiento de losreceptores de retinoides y hormonas tiroideas (SMRT, por Silencing mediator of retinoid and thyroid hormone receptors).10 Por su parte, hasta el momento solo se ha descrito una de las desmetilasas de lisina que actúa a través de cofactores y es específica para los residuos H3K4 dimetilados o monometilados. No se han encontrado desmetilasas de arginina; sin embargo, se observó que una deiminasa de arginina es capaz de desmetilar residuos de arginina monometilados en H3 y H4. Estas enzimas son reguladas por señales emitidas por receptores de estrógenos y andrógenos, lo que sugiere un mecanismo reversible. Las modificaciones de las histonas son procesos dinámicos e íntimamente interrelacionados; por ejemplo, la ubiquitinación de un residuo facilita la fosforilación de otro o impide la metilación de algún otro en una posición diferente y, como es de esperar, la acetilación de un residuo impide su fosforilación.
Otros mecanismos epigenéticos relevantes en la regulación génica son la metilación del ADN que ocurre en las bases de citosina próximas a bases de guanina (regiones CpG) y la impronta genética. En las células somáticas humanas la 5mC (metilcitocina) corresponde al 1%de las bases del ADN, y conforma el 70 a 80%de todas las regiones CpG en el genoma. La metilación puede regular directamente la expresión de un gen al interferir en el momento de la transcripción estimulando la interacción de las proteínas de unión del ADN a los grupos metilo, o indirectamente al cambiar la conformación regional de la cromatina.11 El mantenimiento y control de la metilación en el ADN dependen principalmente de la presencia de las DNMT (DNA–methyltransferases) y de otras metiltransferasas como DNMT 3a y DNMT 3b, que parecen ser más importantes en la etapa embrionaria 12 y tienen una preferencia regional en las islas CpG ubicadas en la porción junto al centrómero.13 La impronta genética se explica por mecanismos de metilación en ciertos genes según su proveniencia paterna o materna11 y este proceso ocurre en la etapa embrionaria del ser humano. En las primeras semanas del desarrollo entre la formación de la mórula y la blástula todo el genoma está sin metilar, de esta manera se facilitan el crecimiento y la diferenciación celulares durante todo el primer trimestre. Luego ocurren la metilación y desacetilación selectivas de regiones CpG y de las histonas, respectivamente. No está totalmente claro por qué la posterior metilación de CpG y la acetilación de histonas ocurren de manera ordenada y no al azar. Sin embargo, se ha demostrado que en la etapa embrionaria los factores nutricionales juegan un papel esencial en este aspecto. Los cambios epigenéticos no solo se limitan al período embrionario, sino que también pueden ocurrir por influencia ambiental en etapas posteriores y la metilación no se limita a los genes relacionados con la impronta ni estos pueden ser modificados solamente en la etapa embrionaria.14
Los microARN constituyen un mecanismo propuesto recientemente como regulador de la expresión génica. Hasta el momento es poco lo que se sabe de estas moléculas; sin embargo, las investigaciones al respecto son cada día más numerosas y el conocimiento sobre ellos se incrementa rápidamente. Los estudios en células humanas y en modelos animales indican que la regulación génica mediada por microARN es importante en el desarrollo humano, la diferenciación celular, la adaptación al ambiente, la oncogénesis y las interacciones de las células huésped con patógenos.15 Existen más de 500 microARN en el genoma cada uno de los cuales puede regular múltiples genes (hasta 200) bloqueando su síntesis proteica mediante la degradación del ARN mensajero.16 Son pocos los estudios sobre los microARN en el asma; no obstante, en un análisis reciente se encontró que un polimorfismo de un solo nucleótido en el gen HLA–G puede afectar la unión de tres microARN a este gen17,18 y alterar la estabilidad de su ARN mensajero. Estos estudios de epidemiología genética sientan las bases para el desarrollo de análisis funcionales in vitro en un futuro cercano.
Es importante anotar que a medida que cada individuo va creciendo y se expone a diferentes influencias ambientales, van aumentando los cambios epigenéticos. Para poder observar las variaciones ocurridas a lo largo del desarrollo de los individuos y las evidencias de que se deben realmente a la influencia ambiental, es necesario estudiar personas con idéntica carga genética. Un ejemplo de este tipo de estudios fue el llevado a cabo por Fraga y colaboradores19 quienes escogieron una cohorte de gemelos monocigotos en un rango de edad entre 3 y 74 años en los que investigaron diferencias en la metilación o acetilación de las histonas. El 65%de los individuos estudiados presentaban contenidos iguales de 5–metilcitocina en el genoma y de histonas 3 y 4 desacetiladas. Sin embargo, el restante 35%tenían diferencias significativas para los dos mecanismos epigenéticos y lo más llamativo fue el hecho de que este grupo lo conformaban las parejas de gemelos de mayor edad. Posteriormente, evaluando la metilación en loci específicos, se encontró que en los gemelos mayores de 50 años eran mayores los cambios en la metilación que en los menores de 28 años con distintos niveles de expresión genética. Los gemelos con menor período de convivencia y los que vivían en condiciones ambientes muy divergentes también presentaban mayores diferencias en la metilación de los loci evaluados y en la expresión de los genes. Estos datos señalan la relevancia de las condiciones ambientales y del tiempo de exposición a los estímulos como reguladores de la expresión génica mediante mecanismos epigenéticos en forma específica. En esta revisión se recopilan las evidencias de la participación de los mecanismos epigenéticos en la inflamación alérgica en el asma además de los estudios epidemiológicos y experimentales indicadores de que el efecto de los factores ambientales en esta enfermedad puede estar mediado, al menos en parte, por estos mecanismos.
MECANISMOS EPIGENÉTICOS EN LA INFLAMACIÓN ALÉRGICA EN ASMA
La importante participación de la epigenética en el asma se empezó a vislumbrar desde los experimentos de inmunohistología en biopsias y lavados broncoalveolares de pacientes con asma leve y moderada en los que se encontró un marcado aumento en la actividad de las acetil–transferasas de histona (HAT) y de la expresión de las enzimas acetiladoras CBP (por, CREB binding protein; CREB, por cAMP response element binding protein, proteína de unión al elemento respondedor de AMP cíclico) y el factor asociado a p300/CREB (PCAF, por p300/ CREB associated factor) y una disminución de las enzimas desacetiladoras HDAC 1–6 lo cual favorece la expresión de genes inflamatorios.20,21 De estos datos se puede concluir que los cambios en la acetilación de las histonas están regulados estrictamente por el buen funcionamiento y expresión de las enzimas que intervienen en ella y que posiblemente la función de estas enzimas se altere en los pacientes asmáticos. Estas diferencias en la expresión parecen ocurrir solo en el pulmón pues no se las ha encontrado al analizar linfocitos y monocitos de sangre periférica de pacientes asmáticos y controles.21 Ya se conocen al nivel molecular algunos de los mecanismos epigenéticos presentes en el proceso inflamatorio del asma y los que se producen como respuesta a medicamentos de uso común como los corticoides. En el tejido bronquial del individuo asmático, se liberan muchas citoquinas como la IL–1 y el TNF–a que transmiten sus señales intracelulares mediante el factor nuclear NF–?B (nuclear factor ?B), el cual también se activa durante las infecciones virales y en otros mecanismos de la respuesta inmune. Este factor tiene particular importancia no solo como un inductor reconocido de la transcripción génica sino como mediador de procesos epigenéticos dado que induce temporalmente la acetilación y otras modificaciones de las histonas22 y recluta otros coactivadores y complejos de remodelación estimulando la expresión de genes inflamatorios.23 En células epiteliales, la acetilación se da principalmente en H4 y está dirigida sobre todo a los residuos K8 y K12 de los elementos reguladores que responden a NF?B.24 El proceso incluye la unión inicial del NF?B al ADN lo que sirve de base para que se reclute un gran complejo coactivador con actividad de acetiltransferasas de histona (HAT) que incluye CREB (cAMP response element binding protein) y PCAF (p300/CREB associated factor) que acetila las histonas adyacentes. Otras HAT que se asocian con el NF?B son GRIP (Glucocorticoid receptor interacting protein–1, proteína de interacción con el receptor de glucocorticoides 1), algunos miembros de la familia p160 y el coactivador–1 del receptor de esteroides (SRC–1 por Steroid receptor coactivator–1).25 La metilación/desmetilación del ADN parece ser otro factor presente en las etapas iniciales de la respuesta alérgica inflamatoria. Se ha observado en estudios in vitro que ocurren cambios cromatínicos durante la diferenciación de los linfocitos hacia un tipo productor Th1 o Th2 y que la desmetilación de los sitios CpG en el promotor y en el primer intrón del gen de la IL– 4 y la hipermetilación del promotor de IFN–?, se asocian con mayor producción de IL–4 y mayor diferenciación Th2. En particular, la metilación de los motivos CpG en la posición –253 del promotor de IFN–? se correlacionó con una polarización de la respuesta hacia el tipo Th2; se dio con la inhibición de la transcripción del gen mediada por un bloqueo de unión de los factores activadores de la trascripción CREB y ATF2/c–jun (Factor activador de la transcripción / protooncogén Jun) a los sitios AP1 de su promotor.26 Como se describe a continuación, estas observaciones se relacionan directamente con los efectos inducidos por algunos factores ambientales como la polución y los alergenos.
FACTORES AMBIENTALES INDUCTORES DE MECANISMOS EPIGENÉTICOS EN EL ASMA
En estudios epidemiológicos longitudinales se ha demostrado que los factores ambientales intrauterinos como la dieta materna, la exposición al humo del cigarrillo, oxidantes, polución, microflora bacteriana y alergenos son de riesgo para alergias y asma y, más aún, que el efecto de agentes como el humo del cigarrillo durante el embarazo puede extenderse hasta la segunda generación lo que sugiere un mecanismo heredable. Otros agentes a los que el individuo está expuesto durante los primeros años de la vida también pueden condicionar un tipo de respuesta protectora o de susceptibilidad para las alergias. Dado que los cambios epigenéticos ocurren principalmente durante esta etapa, se propone que este puede ser el mecanismo subyacente al efecto de estos factores y que, de igual manera, la historia natural característica de la enfermedad se debe a cambios epigenéticos ocurridos durante la infancia, la adolescencia y la vida adulta, que son susceptibles de ser inducidos por factores ambientales.8 Por su parte, algunos investigadores han planteado la hipótesis de que muchos de los genes participantes en la síntesis de IgE y en el proceso de remodelación bronquial, son genes fetales persistentes o reminiscentes, que no se silencian durante la vida in útero o la infancia temprana por mecanismos epigenéticos y que sus productos pueden iniciar y mantener los mecanismos patogénicos en el asma alérgica.27
Humo de cigarrillo
Por medio de estudios epidemiológicos se han documentado ampliamente los efectos de la exposición al humo del cigarrillo, ya sea posnatal o durante la vida intrauterina, sobre el desarrollo y la función pulmonares. Este factor se asocia con un mayor riesgo de tener asma,28 con una merma de la función y del crecimiento pulmonares y con la presencia de sibilancias en la infancia.29 Los niños expuestos al humo del cigarrillo presentan vías aéreas de calibre pequeño con relación a su tamaño corporal, con engrosamiento, incremento del tono del músculo liso, disminución de la elasticidad pulmonar y mayor predisposición a procesos inflamatorios.30 Muchos estudios transversales y de cohorte han observado esta asociación. En un análisis en el que se incluyeron 1.737 niños preescolares australianos se halló que la exposición en la etapa prenatal fue un factor de riesgo para tener sibilancias en el primer año de vida (OR: 1,6; IC 95%: 1,0–2,6), sibilancias en los últimos 12 meses (OR: 1,5; IC 95%: 1,0–2,4) y asma (OR: 2,1; IC 95%: 1,0–4,1).31 Resultados similares se observaron en 5.762 escolares del sur de California (Estados Unidos); en ellos, la exposición al humo del tabaco in útero, pero no en la etapa posnatal, se relacionó con asma (OR: 1,8; IC 95%: 1,1–2,9), asma con síntomas actuales (OR: 2,3; IC 95%: 1,3–4,0) y sibilancias asociadas a infecciones virales (OR: 2,1; IC 95%: 1,3–3,4) y sin infecciones (OR: 2,5; IC 95%: 1,4–4,4) entre otras.29 La exposición en el período posnatal también se ha relacionado fuertemente con una mayor frecuencia de exacerbaciones en niños con asma.32,33 y con la ocurrencia de bronquitis sibilante.34 Por otro lado, se ha demostrado que la exposición in útero está asociada con un nivel alto de IgE en el cordón umbilical de los hijos al nacer,35 mayor linfoproliferación en neonatos36 y una respuesta de células Th1 disminuida en los ensayos de estimulación policlonal in vitro,37 además de merma en la producción de las citoquinas IL–6, IL–10 e IFN–? después de la estimulación de los receptores tipo Toll (Toll like receptors) TLR2, TLR3, TLR4 y TLR9 que son esenciales en la respuesta inmune innata.38 Estos estudios indican modificaciones en varios de los componentes de la respuesta inmune en los hijos de madres fumadoras. Uno de los mecanismos responsables de los efectos del humo del cigarrillo puede ser la inducción de estrés oxidativo y la producción de radicales libres que alteran las respuestas dependientes de oxígeno en los macrófagos y en otras células presentadoras de antígenos y disminuyen la producción de IL–12 tanto en ratones39 como en seres humanos.40 El segundo mecanismo son las modificaciones en la colonización bacteriana de las mucosas con mayor adherencia de los patógenos, además de las alteraciones de la respuesta inmune local y mayor potencia de la penetración alergénica en el epitelio debida al daño producido por las más de 2.000 sustancias irritantes presentes en el humo del tabaco.41 Es de anotar que la exposición al humo del cigarrillo es un ejemplo de cómo la influencia de un factor ambiental es evidente en un contexto genético determinado: el efecto es más evidente en individuos con ciertas variantes en genes de proteínas antioxidantes como es el caso de los hijos de madres fumadoras con el gen nulo de la transferasa de glutatión S (GST, por Glutathione S transferase), los cuales tienen una mayor susceptibilidad a sufrir asma en comparación con los que tienen los alelos protectores.
En análisis in vitro se ha observado la inducción de cambios en moléculas participantes en las modificaciones epigenéticas. En líneas celulares epiteliales como A549 y BEAS–B se demostró que el estrés oxidativo inducido por el humo del cigarrillo acetila las histonas, especialmente la H4 de lo que resulta un incremento de la liberación de citoquinas inflamatorias como IL–8.42 El efecto del estrés oxidativo puede estar dado, al menos en parte, por las semiquinonas, compuestos orgánicos que tienen la capacidad de generar radicales hidroxilo y peróxido de hidrógeno y de esta manera alterar el equilibrio de óxidoreducción (redox) celular. En células epiteliales pulmonares aumenta la transcripción de IL–8 y de otras citoquinas inflamatorias a través de la activación del NF– ?B y la proteína activadora 1(AP–1). Específicamente, se ha observado que el estrés oxidativo aumenta la asociación entre el componente p65 del NF–?B y el coactivador CBP.43 (Figura n.° 1).
Ver (Figura1)
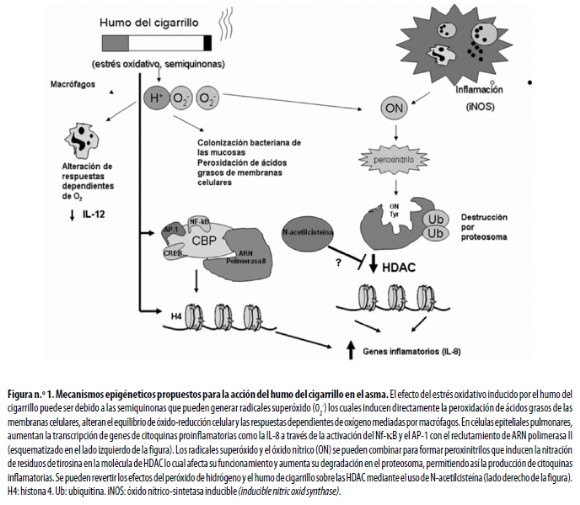
Otros componentes responsables son los radicales hidroxilo, superóxido y óxido nítrico que se encuentran en la fase gaseosa del humo. En forma directa, los iones superóxido e hidroxilo inducen la peroxidación de los ácidos grasos poliinsaturados de la membrana celular alterando la estructura, composición y permeabilidad de la misma con inactivación de los receptores y enzimas, incremento de la permeabilidad tisular y daño pulmonar.44 Por su parte, el superóxido y el óxido nítrico se combinan para formar peroxinitrilos que actúan sobre las desacetilasas de histonas (HDAC). Se considera que la inactivación de las HDAC2 implica la nitración de residuos de tirosina en la molécula lo que teóricamente podría alterar el sitio catalítico de la enzima afectando su eficiencia y aumentando su degradación en el proteosoma como sucede con otras enzimas.45 Se ha demostrado que el peróxido de hidrógeno y el humo condensado de cigarrillo reducen marcadamente la actividad de las desacetilasas de histonas (HDAC) y la expresión de HADC2 en las células epiteliales46 y que esos efectos son reversibles mediante el uso del antioxidante N–acetilcisteína47 (Figura n.° 1). Además de lo anterior, en asmáticos fumadores hay una reducción notoria y significativa de la actividad de las HDAC en el tejido bronquial en comparación con asmáticos no fumadores;48 esta reducción también está asociada con mayor gravedad del asma y menor respuesta a los esteroides.49
Polución ambiental
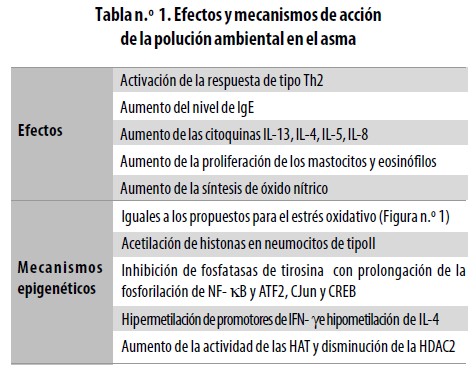
La polución ambiental consiste en diminutas partículas con un diámetro menor de 10–15 µm (PM10) provenientes del polvo, humo o aerosoles líquidos producidos por vehículos y fábricas, quema de madera, sitios de construcción y cultivos con arado.50 Puede incluir también la ceniza residual liberada durante la combustión de aceites de bajo grado, que es una mezcla inorgánica de silicatos y sales metálicas que contiene vanadio, zinc, hierro y níquel.51 Se ha observado en experimentos en animales y en seres humanos y en muchos análisis in vitro que el material particulado y en especial las partículas de diesel inducen una polarización de la respuesta inmune hacia el tipo Th2 y una mayor producción de IgE y citoquinas de este perfil52,53(Tabla n.°1). La exposición a hidrocarburos aromáticos policíclicos como el fenantreno eleva el nivel de IgE en personas fumadoras y alérgicas, así como en individuos sanos no fumadores; además, la exposición a partículas y gases de hidrocarburos aromáticos aun por cortos períodos (4 a 6 horas) puede elevar la producción de citoquinas como IL–3, IL–4, IL–5 e IL8, aumentar la proliferación de mastocitos y eosinófilos y promover la síntesis de óxido nítrico (ON). Estos efectos parecen estar mediados por su acción coadyuvante ante la exposición a aeroalergenos puesto que cuando individuos alérgicos se exponen a alergenos junto con derivados de diesel la respuesta es mayor que cuando se los estimula solo con alergenos, o solo con dichos derivados. Las PM10 pueden fácilmente llegar a las vías aéreas inferiores y generar un proceso inflamatorio por lesión directa del epitelio o interactuando con los receptores de hidrocarburos aromáticos. Se han propuesto otros mecanismos de acción: en células epiteliales humanas estos compuestos activan los genes proinflamatorios mediante los fenómenos inducidos por el estrés oxidativo54 previamente descritos. Además, los hidrocarburos aromáticos también actúan aumentando directamente la acetilación de histonas o actuando sobre las enzimas acetiladoras y desacetiladoras (Tabla n.° 1). Por su parte, el vanadio puede inhibir las fosfatasas de tirosina prolongando la fosforilación de NF–?B y de otros factores de transcripción incluyendo el ATF–2 (Activating transcription factor 2), c–Jun y CREB55 y las partículas de diesel inhaladas y el carbón causan la metilación de los genes p16INK4a56 en tumores de ratas. Un estudio publicado recientemente57 describe la relación entre la exposición a partículas de diesel junto con alergenos de hongos (Aspergillus fumigatus) y una respuesta de IgE aumentada en un modelo murino in vivo. En tal investigación, la exposición a estas sustancias indujo una hipermetilación de los promotores del gen de IFNg y una hipometilación del promotor del gen de IL–4; hubo una correlación fuerte entre el grado de metilación y el nivel sérico de IgE total en los ratones analizados. En estudios in vitro con neumocitos de tipo II, la exposición a material particulado con diámetro aerodinámico menor de 10 µm indujo la remodelación de la cromatina mediante acetilación de histonas58 y en cultivos de células A549 estimulados con PM10, H2O2 o tricostatina A (TSA) la expresión de la IL–8 fue mayor que la observada en los cultivos no estimulados.58 Cuando las células se expusieron por 24 horas simultáneamente a TSA y PM10 o a TSA y H2O2 el nivel de expresión de IL–8 se incrementaba hasta dos veces por encima del encontrado con PM10 o H2O2 solas y hubo una mayor acetilación de la histona 4 cuando las células estuvieron en un medio con PM10 y H2O2, de manera dependiente del nivel de PM10. La actividad de las HAT se encontraba aumentada y el nivel de HDAC2 estaba disminuido en las células que recibieron PM10, H2O2 o TSA.58 Otro grupo demostró, por medio de aislamiento de las proteínas del núcleo, que en las células expuestas a estos elementos había mayor translocación del NF–kB hacia el núcleo.42 Como este, existen varios estudios que señalan el papel de los hidrocarburos aromáticos y de otras partículas inhaladas de PM10 en la acetilación de las histonas de determinados genes, en especial de aquellos con actividad proinflamatoria.22,59
Ver (Tabla1)
Infecciones virales
La infección por adenovirus incrementa la expresión de los genes inflamatorios en las células epiteliales in vitro y esto parece estar mediado por la proteína E1A que interactúa con los coactivadores con actividad HAT como los CBP.60 En estudios en curíes se ha observado que la infección por adenovirus amplifica la respuesta inflamatoria a alergenos 61 y se asocia con una reducción en la actividad de las desacetilasas de histonas (HDAC) en los animales sensibilizados con ovoalbúmina. La persistencia de las infecciones por adenovirus también se ha asociado con asma infantil resistente al tratamiento con esteroides.61
MECANISMOS EPIGENÉTICOS EN LA TERAPIA CON CORTICOIDES Y TEOFILINA
Los corticoides son medicamentos antiinflamatorios muy efectivos y ampliamente utilizados en el tratamiento del asma y otras enfermedades crónicas. Su principal efecto es disminuir la producción de múltiples proteínas proinflamatorias como citoquinas, quimioquinas, moléculas de adhesión, enzimas inflamatorias, receptores y proteínas que han sido activadas durante la inflamación. La inhibición de la transcripción se da principalmente revirtiendo la acetilación de histonas; el complejo conformado por el corticoide y su receptor se une a coactivadores y reclutan HDAC2 en los sitios de transcripción activa.62 Esto explica parcialmente la resistencia a corticoides observada en el asma grave en la que están disminuidas la expresión y actividad de las desacetilasas de histonas (HDAC). En mayores concentraciones, los receptores forman homodímeros que interactúan con sus sitios de reconocimiento del ADN para activar la transcripción de genes antiinflamatorios como la anexina–1 (lipocortina–1), IL–10 y el inhibidor de NF–?B (I?B–a). Para hacer esto, los receptores activados se acoplan a moléculas coactivadoras con actividad de acetil–transferasas de histonas (HAT) como la CBP o proteína de unión a CREB (CREB binding protein), el coactivador del receptor de esteroides–1 (SRC–1, Steroid receptor coactivator–1), la proteína 1 que interactúa con el receptor de glucocorticoides GRIP–1 (Glucocorticoid receptor interacting protein–1) y el factor asociado a p300/ CBP (pCAF).63 La activación está asociada también con acetilación de los residuos de lisina 5 y 6 en H4.24 Con dosis terapéuticas se inhibe también la transcripción de varios genes relacionados con los efectos colaterales tales como el de la propiomelanocortina y el factor liberador de corticotropina, osteocalcina y queratinas.62 Por su parte, la teofilina, un broncodilatador de amplio uso en pacientes con asma también altera los mecanismos epigenéticos en un efecto independiente de su función como inhibidor de las fosfodiesterasas. En experimentos con células epiteliales y macrófagos in vitro y modelos murinos in vivo se demostró que dosis bajas de teofilina aumentan la actividad de las desacetilasas de histonas (HDAC) las cuales pueden entonces servir de blanco de acción para los corticoides.64 El efecto en las desacetilasas de histonas se da a concentraciones terapéuticas (10–6 a 10–5 M) pero no a altas concentraciones (10–4 M) y es bloqueado por la tricostatina, un inhibidor de la actividad de dichas desacetilasas. El efecto directo sobre estas enzimas se observó también en biopsias bronquiales de pacientes asmáticos en los que aumentó su actividad después del tratamiento con dosis bajas de teofilina (concentración media plasmática de 5 mg/L). Mediante su acción sobre las HDAC, la teofilina también es capaz de revertir el efecto del estrés oxidativo y del humo del cigarrillo así como de restablecer o potenciar la respuesta a corticoides en individuos con asma grave y en fumadores. Esto explica al nivel molecular el efecto sinérgico observado con el tratamiento combinado en estos pacientes.65
Otros factores
Otros factores que pudieran influir en el riesgo de asma en la infancia incluyen la ingesta materna de frutas, legumbres y aceites de pescados,66 alimentos que contienen vitaminas, minerales, probióticos67 o el uso de antibióticos durante el embarazo.68Algunos estudios epidemiológicos sustentan que con el consumo materno de alimentos o suplementos ricos en vitamina E o zinc, durante el primer y segundo trimestres del embarazo, disminuye la incidencia de sibilancias en los niños menores de dos años.69 El consumo de estos productos en los pacientes también influye en los signos y síntomas de la enfermedad. En niños italianos entre 6–8 años que consumían durante el invierno una dieta rica en frutas cítricas, con alto nivel de vitamina C, se observó una menor incidencia de sibilancias que en el grupo control.70 En una población irlandesa se evaluó si niveles altos de vitaminas C, E y ß–caroteno se asociaban a disminución de síntomas respiratorios como la tos, las sibilancias, las crisis asmáticas, etc. Los investigadores observaron que no había una mejoría significativa en los síntomas clínicos, pero que los resultados del volumen espiratorio forzado (FEV) y la capacidad vital forzada (CVF) eran mejores en quienes tenían un consumo alto de vitaminas y âcaroteno. 71 Otros autores,72 para evaluar el efecto la vitamina D en el asma, utilizaron ratones knock–out (o sea, con uno o más genes inactivados) para el receptor de la vitamina D y ratones silvestres a los cuales se les indujo asma por medio del reto con ovoalbúmina y se les administró 1,25–[OH]2–D3. En los ratones silvestres se produjeron síntomas respiratorios, hiperreactividad bronquial, infiltración de neutrófilos y elevación de citoquinas del tipo Th2 y no hubo cambios en la intensidad de la respuesta asmática después de la administración de la vitamina D, pero sí se observó un incremento en la expresión de dos genes relacionados con la respuesta Th2 (sCD23 y GATA–3). Por su parte en los ratones knock–out, aunque no se produjo una respuesta inflamatoria o de hipersensibilidad bronquial, sí se observaron concentraciones más altas de IgE y de citoquinas de tipo Th2. Esto hace pensar que la interacción de la vitamina D con su ligando, que se puede encontrar en la región promotora de varios genes, juega un papel clave en el desarrollo del asma afectando la expresión de los genes de las citoquinas Th2. En otros experimentos con ratones silvestres a los que se les administraron diferentes dosis de vitamina D73 se observó aumento en la expresión de IL–4 e IL–13 y mayor proliferación de los linfocitos de tipo Th2; sin embargo, el número de eosinófilos fue menor en los lavados broncoalveolares y el nivel de IL–5 fue menor en comparación con el de ratones de control no inoculados. En conjunto estos experimentos muestran que la vitamina D juega un papel dual en la respuesta de tipo Th2 tanto estimulando como disminuyendo la proliferación y expresión de células y citoquinas características de este perfil. Esto lo hace por interacción directa con distintos factores de transcripción y regiones promotoras de diferentes genes. La influencia sobre el factor de transcripción GATA–3 (importante en la regulación de la transcripción de las citoquinas IL–4, IL–5 e IL–13) pudiera explicar en parte la elevación de la IL–4 y la IL–13 en los ratones que reciben una dieta rica en este metabolito. En resumen, aunque falta mucho por explorar, algunos estudios in vitro y en animales permiten empezar a vislumbrar el posible mecanismo de acción de factores dietéticos en el desarrollo del asma y otras enfermedades alérgicas en los seres humanos.
CONCLUSIÓN
Los estudios revisados muestran cómo el efecto de los factores ambientales y de la inflamación en el asma está mediado por una serie de mecanismos epigenéticos que alteran tanto el riesgo de padecer esta enfermedad como su curso. Esto resalta la plasticidad del genoma y su regulación en una respuesta emergente a estos factores. El estudio de tales mecanismos es un campo de investigación abierto, novedoso e interesante cuyos resultados incrementarán el conocimiento sobre las variaciones del epigenoma potencialmente heredables pero también adquiridas, modulables y susceptibles de ser utilizadas en estrategias terapeúticas en asma, alergias y otras enfermedades inflamatorias crónicas.
REFERENCIAS BIBLIOGRÁFICAS
1. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 2008; 8: 183–192. [ Links ]
2. Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol 2008; 8: 205–217. [ Links ]
3. Vercelli D. Discovering susceptibility genes for asthma and allergy. Nat Rev Immunol 2008; 8: 169–182. [ Links ]
4. Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 2002; 418: 426–430. [ Links ]
5. Laitinen T, Daly MJ, Rioux JD, Kauppi P, Laprise C, Petays T, et al. A susceptibility locus for asthma–related traits on chromosome 7 revealed by genome–wide scan in a founder population. Nat Genet 2001; 28: 87–91. [ Links ]
6. Cheung P, Lau P. Epigenetic regulation by histone methylation and histone variants. Mol Endocrinol 2005; 19: 563–573. [ Links ]
7. Adcock IM, Ford P, Ito K, Barnes PJ. Epigenetics and airways disease. Respir Res 2006; 7: 21–40. [ Links ]
8. Miller RL, Ho SM. Environmental epigenetics and asthma: current concepts and call for studies. Am J Respir Crit Care Med 2008; 177: 567– 573. [ Links ]
9. Rice JC, Allis CD. Code of silence. Nature 2001; 414: 258– 261. [ Links ]
10. Privalski ML. The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annu Rev Physiol 2004; 66: 315–360. [ Links ]
11. Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition 2004; 20: 63–68. [ Links ]
12. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16: 6–21. [ Links ]
13. Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, et al. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000; 404: 1003–1007. [ Links ]
14. Waterland RA, Garza C. Early postnatal nutrition determines adult pancreatic glucose–responsive insulin secretion and islet gene expression in rats. J Nutr 2002; 132: 357–364. [ Links ]
15. Boyd SD. Everything you wanted to know about small RNA but were afraid to ask. Lab Invest 2008; 88: 569–578. [ Links ]
16. Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet 2005; 37: 495–500. [ Links ]
17. Hunt JS, Petroff MG, McIntire RH, Ober C. HLA–G and immune tolerance in pregnancy. FASEB J 2005; 19: 681– 693. [ Links ]
18. Tan Z, Randall G, Fan J, Camoretti–Mercado B, Brockman– Schneider R, PanL, et al. Allele–specific targeting of microRNAs to HLA–G and risk of asthma. Am J Hum Genet 2007; 81: 829–834. [ Links ]
19. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 2005; 102: 10604–10609. [ Links ]
20. Ito K, Caramori G, Lim S, Oates T, Chung KF, Barnes PJ, et al. Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med 2002; 166: 392–396. [ Links ]
21. Cosio BG, Mann B, Ito K, Jazrawi E, Barnes PJ, Chung KF, et al. Histone acetylase and deacetylase activity in alveolar macrophages and blood monocytes in asthma. Am J Respir Crit Care Med 2004; 170: 141–147. [ Links ]
22. Lee KY, Ito K, Hayashi R, Jazrawi EP, Barnes PJ, Adcock IM. NF–kappaB and activator protein 1 response elements and the role of histone modifications in IL–1beta–induced TGF–beta1 gene transcription. J Immunol 2006; 176: 603– 615. [ Links ]
23. Ghosh S, Karin M. Missing pieces in the NF–kappaB puzzle. Cell 2002; 109 (Suppl.): S81–S96. [ Links ]
24. Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin– 1beta–induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000; 20: 6891–6903. [ Links ]
25. Jenkins BD, Pullen CB, Darimont BD. Novel glucocorticoid receptor coactivator effector mechanisms. Trends Endocrinol Metab 2001; 12: 122–126. [ Links ]
26. Jones B, Chen J. Inhibition of IFN–? transcription by sitespecific methylation during T helper cell development. EMBO J 2006; 25: 2443–2452. [ Links ]
27. Bousquet J, Jacot W, Yssel H, Vignola AM, Humbert M. Epigenetic inheritance of fetal genes in allergic asthma. Allergy 2004; 59: 138–147. [ Links ]
28. Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. The Group Health Medical Associates. N Engl J Med 1995; 332: 133–138. [ Links ]
29. Gilliland FD, Li YF, Peters JM. Effects of maternal smoking during pregnancy and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med 2001; 163: 429–436. [ Links ]
30. Tepper RS, Williams–Nkomo T, Martinez T, Kisling J, Coates C, Daggy J. Parental smoking and airway reactivity in healthy infants. Am J Respir Crit Care Med 2005; 171: 78–82. [ Links ]
31. Horak E, Morass B, Ulmer H. Association between environmental tobacco smoke exposure and wheezing disorders in Austrian preschool children. Swiss Med Wkly 2007; 137: 608–613. [ Links ]
32. Weitzman M, Gortmaker S, Walker DK, Sobol A. Maternal smoking and childhood asthma. Pediatrics 1990; 85: 505–511. [ Links ]
33. Chilmonczyk BA, Salmun LM, Megathlin KN, Neveux LM, Palomaki GE, Knight GJ, et al. Association between exposure to environmental tobacco smoke and exacerbations of asthma in children. N Engl J Med 1993; 328: 1665–1669. [ Links ]
34. Wright AL, Holberg C, Martinez FD, Taussig LM. Relationship of parental smoking to wheezing and nonwheezing lower respiratory tract illnesses in infancy. Group Health Medical Associates. J Pediatr 1991; 118: 207–214. [ Links ]
35. Magnusson CG. Maternal smoking influences cord serum IgE and IgD levels and increases the risk for subsequent infant allergy. J Allergy Clin Immunol 1986; 78: 898–904. [ Links ]
36. Devereux G, Barker RN, Seaton A. Antenatal determinants of neonatal immune responses to allergens. Clin Exp Allergy 2002; 32: 43–50. [ Links ]
37. Noakes PS, Holt PG, Prescott SL. Maternal smoking in pregnancy alters neonatal cytokine responses. Allergy 2003; 58: 1053–1058. [ Links ]
38. Noakes PS, Hale J, Thomas R, Lane C, Devadason SG, Prescott SL. Maternal smoking is associated with impaired neonatal toll–like–receptor–mediated immune responses. Eur Respir J 2006; 28: 721–729. [ Links ]
39. Murata Y, Shimamura T, Hamuro J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int Immunol 2002; 14: 201–212. [ Links ]
40. Utsugi M, Dobashi K, Ishizuka T, Endou K, Hamuro J, Murata Y, et al. c–Jun N–terminal kinase negatively regulates lipopolysaccharide–induced IL–12 production in human macrophages: role of mitogen–activated protein kinase in glutathione redox regulation of IL–12 production. J Immunol 2003; 171: 628–635. [ Links ]
41. Prescott SL. Effects of early cigarette smoke exposure on early immune development and respiratory disease. Paediatr Respir Rev 2008; 9: 3–9. [ Links ]
42. Tomita K, Barnes PJ, Adcock IM. The effect of oxidative stress on histone acetylation and IL–8 release. Biochem Biophys Res Commun 2003; 301: 572–577. [ Links ]
43. Rahman I, Marwick J, Kirkham P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF–kB and pro–inflammatory gene expression. Biochem Pharmacol 2004; 68: 1255–1267. [ Links ]
44. Rahman I. Oxidative stress, transcription factors and chromatin remodelling in lung inflammation. Biochem Pharmacol 2002; 64: 935–942. [ Links ]
45. Schmidt P, Youhnovski N, Daibert A, Balan A, Arsic M, Bachschmidt M, et al. Specific nitration at tyrosine 430 revealed by high resolution mass spectrometry as basis for redox regula tion of bovine prostacyclin synthase. J Biol Chem 2003; 278: 12813–12819. [ Links ]
46. Ito K, Tomita T, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase (HDAC)2 activity and enhances IL–8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun 2004; 315: 240–245. [ Links ]
47. Moodie FM, Marwick JA, Anderson CS. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF–kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J 2004; 18: 1897–1899. [ Links ]
48. Murahidy A, Ito M, Adcock IM, Barnes PJ, Ito K. Reduction in histone deacetylase expression and activity in smoking asthmatics; a mechanism of steroid resistance. Proc Am Thorac Soc 2005; 2: A889. [ Links ]
49. Thomson NC, Chaudhuri R, Livingston E. Active cigarette smoking and asthma. Clin Exp Allergy 2003; 33: 1471– 1475. [ Links ]
50. Edwards T, Peterson–Myers J. Environmental exposures and gene regulation in disease etiology. Environ Health Perspect 2007; 115: 1264–1270. [ Links ]
51. Henry WM, Knapp KT. Compound forms of fossil fuel flyash emissions. Environ Sci Technol 1980; 14: 450–456. [ Links ]
52. Bommel H, Li–Weber M, Serfling E, Duschl A. The environmental pollutant pyrene induces the production of IL–4. J Allergy Clin Immunol 2000; 105: 796–802. [ Links ]
53. Fujieda S, Diaz–Sanchez D, Saxon A. Combined nasal challenge with diesel exhaust particles and allergen induces in vivo IgE isotype switching. Am J Respir Cell Mol Biol 1998; 19: 507–512. [ Links ]
54. Gilmour PS, Rahman I, Hayashi S, Hogg JC, Donaldson K, McNee W. Adenoviral E1A primes alveolar epithelial cells to PM10–induced transcription of interleukin–8. Am J Physiol Lung Cell Mol Physiol 2001; 281: L598–L606. [ Links ]
55. Samet JM, Silbajoris R, Huang T, Jaspers I. Transcription factor activation following exposure of an intact lung preparation to metallic particulate matter. Environ Health Perspect 2002; 110: 985–990. [ Links ]
56. Belinsky SA, Snow SS, Nikula KJ, Finch GL, Tellez CS, Palmisano WA. Aberrant CpG island methylation of the p16(INK4a) and estrogen receptor genes in rat lung tumors induced by particulate carcinogens. Carcinogenesis 2002; 23: 335–339. [ Links ]
57. Liu J, Ballaney M, Al–alem U, Quan C, Jin X, Perera F, et al. Combined inhaled diesel exhaust particles and allergen exposure alter methylation of T helper genes and IgE production in vivo. Toxicol Sci 2008; 102: 76–81. [ Links ]
58. Gilmour PS, Rahman I, Donaldson K, MacNee W. Histone acetylation regulates epithelial IL–8 release mediated by oxidative stress from environmental particles. Am J Physiol Lung Cell Mol Physiol 2003; 284: L533–L540. [ Links ]
59. Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, et al. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 2004; 31: 633–642. [ Links ]
60. Higashimoto Y, Elliott WM, Behzad AR, Sedgwick EG, Takei T, Hogg JC, et al. Inflammatory mediator mRNA expression by adenovirus E1A–transfected bronchial epithelial cells. Am J Respir Crit Care Med 2002; 166: 200–207. [ Links ]
61. Yamada K, Elliott WM, Brattsand R, Valeur A, Hogg JC, Hayashi S. Molecular mechanisms of decreased steroid responsiveness induced by latent adenoviral infection in allergic lung inflammation. J Allergy Clin Immunol 2002; 109: 35–42. [ Links ]
62. Barnes PJ. How corticosteroids control inflammation. Br J Pharmacol 2006; 148: 245–254. [ Links ]
63. Kurihara I, Shibata H, Suzuki T, Ando T, Kobayashi S, Hayashi M, et al. Expression and regulation of nuclear receptor coactivators in glucocorticoid action. Mol Cell Endocrinol 2002; 189: 181–189. [ Links ]
64. Ito K, Lim S, Caramori G, Cosio B, Fan Chung K, Adcock I, et al. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. PNAS 2002; 99: 8921– 8926. [ Links ]
65. Barnes PJ. Theophylline. New perspectives for an old drug. Am J Respir Crit Care Med 2003; 167: 813–818. [ Links ]
66. Fitzsimon N, Fallon U, O'Mahony D, Loftus BG, Bury G, Murphy AW, et al. Mothers' dietary patterns during pregnancy and risk of asthma symptoms in children at 3 years. Ir Med J 2007; 100 (Suppl.): 27–32. [ Links ]
67. Kukkonen K, Savilahti E, Haahtela T, Juntunen–Backman K, Korpela R, Poussa T, et al. Probiotics and prebiotic galactooligosaccharides in the prevention of allergic diseases: a randomized, double–blind, placebo–controlled trial. J Allergy Clin Immunol 2007; 119: 192–198. [ Links ]
68. Jedrychowski W, Galas A, Whyatt R, Perera F. The prenatal use of antibiotics and the development of allergic disease in one year old infants: a preliminary study. Int J Occup Med Environ Health 2006; 19:707–716. [ Links ]
69. Litonjua AA, Rifas–Shiman SL, Ly NP, Tantisira KG, Rich– Edwards JW, Camargo CA, Jr., et al. Maternal antioxidant intake in pregnancy and wheezing illnesses in children at 2 years of age. Am J Clin Nutr 2006; 84: 903–911. [ Links ]
70. Forastiere F, Pistelli R, Sestini P, Fortes C, Renzoni E, Rusconi F, et al. Consumption of fresh fruit rich in vitamin C and wheezing symptoms in children. SIDRIA Collaborative Group, Italy (Italian Studies on Respiratory Disorders in Children and the Environment). Thorax 2000; 55: 283–288. [ Links ]
71. Grievink L, Smit HA, Ocke MC, van 't Veer P, Kromhout D. Dietary intake of antioxidant (pro)–vitamins, respiratory symptoms and pulmonary function: the MORGEN study. Thorax 1998; 53: 166–171. [ Links ]
72. Wittke A, Weaver V, Mahon BD, August A, Cantorna MT. Vitamin D receptor–deficient mice fail to develop experimental allergic asthma. J Immunol 2004; 173: 3432–3436. [ Links ]
73. Matheu V, Back O, Mondoc E, Issazadeh–Navikas S. Dual effects of vitamin D–induced alteration of TH1/TH2 cytokine expression: enhancing IgE production and decreasing airway eosinophilia in murine allergic airway disease. J Allergy Clin Immunol 2003; 112: 585–592. [ Links ]
Recibido: septiembre 08 de 2008
Aceptado: junio 21 de 2008

