










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.23 no.1 Medellín Jan./Mar. 2010
INVESTIGACIÓN ORIGINAL
Comportamiento de la mutación mtDNA A3243G en dos familias antioqueñas de pacientes diagnosticados con el síndrome MELAS
Behavior of the mtDNA mutation A3243G in two antioquian families of patients with melas syndrome
María Victoria Parra Marín1; Jose William Cornejo Ochoa2; Constanza Elena Duque Vélez1; Andrés Ruíz Linares3; Gabriel Bedoya Berrío4
1 Bióloga, MSc, Universidad de Antioquia
2 Neurólogo Clínico y Epidemiólogo; docente del Departamento de Pediatría y Puericultura, Facultad de Medicina, Universidad de Antioquia
3 Médico, PhD, University College London
4 Licenciado en Biología y Química, Profesor titular, Universidad de Antioquia Correspondencia: Maria Victoria Parra Marín dqvicky@yahoo.com
RESUMEN
Introducción: mutaciones en mtDNA causan citopatias mitocondriales, la más común de ellas es el síndrome MELAS; la transición A3243G en tRNA de leucina (tRNALeu) se presenta en 80% de pacientes. La heteroplasmia, observada en citopatias mitocondriales, consiste en coexistencia de moléculas mutadas y normales en una célula, situación en la cual, dependiendo de su cantidad,afecta su función con expresión clínica variable.
Objetivo: evaluar el comportamiento de la cantidad de heteroplasmia de la mutación 3243G en su expresión clínica y en la dependencia de variantes nucleares.
Pacientes y métodos: se buscaron mutaciones en el gen que codifica para el tRNA de leucina por secuencia y por PCR–RFLP en 34 pacientes, y se tamizó en familiares de los portadores de la mutación. Se tipificaron cuatro Specific Population Allele (SPA) en pacientes y familiares con la mutación A3243G.
Resultados: se halló la mutación A3243G en el tRNALeu en dos pacientes, luego de tamizar la mutación A3243G en ambas familias se evaluó la cantidad de mtDNA mutado (MDNA),encontrando que los casos índices de ambas familias esentaron la mayor cantidad de MDNA; en la primera familia se detectó la mutación en 15 miembros que presentaron diversos síntomas. En la segunda familia se detectó la mutación en un miembro con parálisis cerebral, en dos con migraña y en uno asintomático.
Conclusiones: la severidad de los síntomas se correlaciona con la cantidad de MDNA, se encontróademás correlación entre mtDNA mutado (MDNA) y el Índice de Ancestría Amerindio en cada individuo (IAA), indicando una posible influencia del contexto nuclear amerindio en la segregación y replicación mitocondrial.
Palabras clave
Apoplejía, Convulsiones, Epilepsia, Heteroplasmia, MELAS, Migraña, Mutación, mtDNA,
SUMMARY
Mitochondrial DNA mutations cause mitochondrial cytopathies. Among them Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke–like episodes (MELA) is the commonest. The transition 3243A>G in the Leucine tRNA is present in 80% of the patients. Heteroplasmy is observed in mitochondrial cytopathies, characterized by the coexistence of mutant and wild type molecules in a cell. Depending on the level of heteroplasmy, function and clinical manifestations might result affected.
Objective: To test the degree of heteroplasmy of the mutation 3243G on its expression (syntoms) and nuclear–variants dependence.
Patients and methods: Mutations in the tRNALeu gene were sought in 34 patients by sequencing and PCR–RFLP. Four SPA (specific population alleles) were typed in patients and their relatives carrying the mutation 3243A>G.
Results: The mutation 3243A>G in the Leucine tRNA gene was found in two patients. This mutation was screened in their relatives and the amount of mutant DNA (MDNA) was assessed. The index cases presented with the higher amounts of MDNA in both families. In family one, the mutation was detected in 14 members, three of which presented with short stature, one with hearing loss, one with type 2 diabetes, 8 with migraine and one healthy individual. In family two the mutation was detected in one member with brain paralysis, two with migraine and one healthy individual.
Conclusions: Severity of the symptoms in patients affected with MELAS is correlated with the amount of MDNA. Furthermore, it was found a correlation between MDNA and IAA, suggesting a possible effect of amerind nuclear ontext in The mitochondrial egregation and replication.
Keywords
Stroke, Heteroplasmy, MELAS, Migraine, Mutation, mtDNA, Epilepsy
INTRODUCCIÓN
La mitocondria es una organela citoplasmática de células eucarionte que produce energía en forma de ATP para la actividad celular,1 proceso que se lleva a cabo a través del ciclo de Krebs, cadena respiratoria (CR) o cadena de transporte de electrones (ETC, por sus siglas del inglés: Electron Transport Chain), y la fosforilación oxidativa (OXPHOS, por su siglas del inglés oxidative phosphorylation), y participa además en procesos de apoptosis y señalización celular.2,3 Posee< dos membranas, una externa lisa y otra interna plegada, formando crestas, donde están ubicados los complejos enzimáticos I al IV (ETC) y el complejo V, donde se lleva a cabo la OXPHOS, complejos que son codificados por el genoma nuclear (nDNA) y el DNA de la mitocondria (mtDNA);2 este último, igual que las demás moléculas necesarias para la biosíntesis mitocondrial, está contenido por la matriz.4 Cuando< se altera la función mitocondrial se afecta la síntesis de ATP y se puede originar un tipo de trastorno del metabolismo oxidativo mitocondrial conocido como cistopatía mitocondrial,5 caracterizado principalmente por presentar afección multisistémica;5–7 debido al doble control genético al que está sometido esta organela1 dicho trastorno puede originarse por mutaciones en el nDNA o en el mtDNA.8
El mtDNA se identificó en la década de los 60, y en los humanos fue secuenciado en 1981;9 es una molécula circular de doble cadena de 16.569 pares de bases,4,9 presente en cientos de copias por célula;1 no posee intrones, codifica para 22 RNA de transferencia (tRNA), 2 RNA ribosomales (rRNA) y 13 polipéptidos2,4 y tiene una tasa mutacional diez veces mayor que la del nDNA.10–12 Aunque es heredado de la madre,11, 13 existen algunos reportes de herencia mitocondrial paterna;14,15 la cantidad de mitocondrias en una célula dependerá de su requerimiento energético.2
Durante la división celular, las mitocondrias se distribuyen al azar entre las células hijas, y por ende, sus genomas,11 originando dos situaciones: que todas la moléculas en una célula o tejido sean del mismotipo (homoplasmia), o que sean de dos tipos diferentes (normales y mutadas) (heteroplasmia).1,16 El fenotipo de una célula heteroplásmica dependerá del porcentaje de mtDNA mutado que posea, con posibles variaciones que van desde menos del 1%, situación en la que se generaría una complementación de la función por parte de las moléculas normales, hasta más del 95%, cuando no se logran suplir las necesidades energéticas y se presenta el fenotipo anormal.7 Esta segregación al azar de las mitocondrias ha sido reevaluada en diversos trabajos en los que se han asociado genes nucleares que actúan de manera cuantitativa (QTLs, por sus siglas del inglés: Quantitative Trait Locus) para dirigir la segregación, por lo cual el grado de heteroplasmia puede depender del contexto genético nuclear en que se encuentre la mitocondria.17, 18
Las citopatías mitocondriales constituyen un variado grupo de trastornos causados por deficiencias en la producción de ATP por parte de la mitocondria,5,7,19–22 que se caracterizan por presentar heterogeneidad fenotípica –clínica–,19,22 endofenotípica –bioquímica–,5 y genética.23,24 Las mutaciones pueden agruparse en dos categorías: reorganizaciones grandes, y con mayor frecuencia, mutaciones puntuales;20,25 cada vez es mayor el reporte de mutaciones en mtDNA asociadas a alguna condición patológica,7, 19 las que pueden ocurrir en rRNA, tRNA, o en genes que codifican para proteínas de los complejos de CR y OXPHOS. Cerca del 58% de las mutaciones puntuales patogénicas en mtDNA reportadas se presenta en tRNAs26, y una de las más frecuentes es la A3243G en el gen tRNALeu, responsable de un síndrome caracterizado por episodios de encefalomielopatia mitocondrial, acidosis láctica, y episodios similares al stroke (síndrome MELAS, por sus siglas del inglés: Mitochondrial Encephalomyopathy, Lactic Acidosis and Strokelike episodes) condición identificada en 1984;27 dado el gran número de mutaciones que se presenta en este gen en individuos no relacionados, ha sido considerado como 'punto caliente' de mutaciones.28
El síndrome MELAS, descrito por Pavlakis en 1984,27 es generalmente de herencia materna y se caracteriza por encefalomiopatía, acidosis láctica, accidentes cerebrovasculares (stroke) y/o fibras rojas rasgadas en tejido muscular (RRF por sus siglas del inglés: Raggedred fibres).19 Puede acompañarse, además, de cefaleatipo migraña, vómito recurrente, debilidad muscular y estatura corta.21 Fue la primera citopatía asociada con una mutación puntual en mtDNA, consecuente con una transición de Adenina 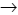 Guanina (AG) en la posición 3243, en la región que codifica para el tRNALeu;19,27 esta transición es responsable del 80% de los casos de MELAS5,7,19 y presenta una gran heterogeneidad genética.30–32
Guanina (AG) en la posición 3243, en la región que codifica para el tRNALeu;19,27 esta transición es responsable del 80% de los casos de MELAS5,7,19 y presenta una gran heterogeneidad genética.30–32
El tRNALeu está codificado entre los nucleótidos 3230 y 3304, y contiene el factor de terminación de la transcripción mitocondrial (mTERF–, por sus siglas del inglés: Mitochondrial transcription termination factor–[nt3229– nt3256]), necesario para la terminación del transcrito ribosomal 16S; es flanqueado por los genes que codifican para el rRNA 16S (1671–3229) y el gen de la NADH deshidrogenasa I33 (3307–4262)24. Todas las mutaciones asociadas con síndrome MELAS son heteroplásmicas, con más del 80% del mtDNA mutado en músculo; la expresión, dependiente de las proporciones de genoma mutante en músculo y cerebro, se hace manifiesta cuando este porcentaje excede el 60%.5,7,19,34 El mtDNA mutado en células sanguíneas es difícil de detectar, pues por la alta proliferación celular su cantidad en ellas es muy escasa.35–37 El efecto de A3243G en la función mitocondrial se produce básicamente en las subunidades con mayor contenido de residuos de Leucina (MT–ND3 y MT–ND6) del complejo proteico I de la CR, situación evidenciada por la deficiencia que este complejo ha mostrado en pacientes con la mutación.38 Una deficiencia en el complejo proteico I podría disminuir el gradiente electroquímico a través de la membrana mitocondrial interna y llevar a una disminución de la respiración y de la síntesis de ATP.28
El mecanismo molecular que puede explicar el síndrome MELAS, como efecto de la transición A3243G, no ha sido dilucidado totalmente, sin embargo, por acercamientos con células cibridas,34 se evidencian las implicaciones de dicha mutación sobre la síntesis de proteínas, dadas por cambios conformacionales del tRNALeu(UUR) mutado, entre las cuales se encuentra la perdida de la especificidad de la Leucil–tranferasa,39–42 que conlleva a la sustitución de leucina por fenilalanina en las proteínas mitocondriales, o a una terminación prematura de la traducción43 La disminución en la tasa de síntesis de proteína (entre 20 – 40%) como efecto de la reducción en el número de ribosomas es otromecanismo que se ha dilucidado.44 Se han realizado numerosos trabajos en los que se observa asociación de esta mutación con otros trastornos diferentes del síndrome MELAS, como sordera45,46 diabetes mellitus tipo 231,46,47,48 y migraña.49,50 Esta variabilidad en la presentación clínica de la mutación puede ser debida a la cantidad de heteroplasmia o al contexto genético nuclear.
En este estudio nos proponemos describir las características clínicas y la correlación genotipofenotipo de dos familias colombianas con la mutación MELAS, y evaluar el efecto del contexto genético nuclear en la segregación mitocondrial.
MATERIALES Y MÉTODOS
Depuración del fenotipo
Se incluyeron pacientes remitidos al laboratorio de Genética Molecular de la Universidad de Antioquia por neurólogos de diferentes instituciones de la ciudad y por los autores, que cumplieran por lo menos con dos de los siguientes criterios:21
– Episodios parecidos a apoplejía (hemiparesia, hemianopsia, ceguera cortical y afasia), con Tomografía Axial Computarizada (TAC) y/o Resonancia magnética nuclear (RMN) que evidenciaran anormalidades focales, principalmente occipitales o en ganglios basales.
– Acidosis láctica en sangre y/o líquido céfaloraquídeo, y/o fibras rojas rasgadas en biopsia muscular.
– Convulsiones generalizadas o focales, demencia, vómito y cefalea recurrente.
–Asociaciones de síntomas y signos descritos en la literatura relacionados con síndrome MELAS, tales como: talla baja, intolerancia al ejercicio, sordera, bloqueo cardiaco, ataxia cerebelosa, atrofia óptica, oftalmoplejia externa progresiva, retinitis pigmentaria, diabetes mellitus tipo 2 de herencia materna, migraña y hemiplejía.
Se reunieron 34 pacientes que cumplieron con los criterios de inclusión antes citados, a quienes se les extendieron las respectivas genealogías con el fin de incluir familiares que presentarán algunos de los síntomas anteriores.
Muestras
Previo consentimiento informado se tomaron muestras de sangre, mucosa oral y/o músculo a cada individuo, y se recolectó un volumen de 5 mL de sangre en tubos VACUTAINER® con EDTA; la mucosa oral se obtuvo empleando enjuagues bucales con 10 mL de solución salina al 0,9%, y la muestra de músculo por biopsia del deltoides, dividida en dos partes, una de las cuales se envió al laboratorio de Patología y otra al de Genética Molecular (GENMOL) de la Universidad de Antioquia; Todas las muestras fueron almacenadas a –20 °C hasta que fueran procesadas.
Análisis bioquímico e histológico
A 12 de los 34 pacientes (35,3%) se les evaluó el nivel de lactato, y a 4 (11,8%) se les realizó estudio histológico para búsqueda de fibras rojas rasgadas. A 12 pacientes (35%) se les evalúo el nivel de lactato y a 4 (11,7%) se les realizó estudio histológico para búsqueda de fibras rojas rasgadas.
Genotipificación
En todas las muestras se extrajo el DNA utilizando el método modificado de fenol–cloroformo. Para identificar mutaciones en el gen mitocondrial que codifica para el tRNALeu se amplificó la región de mtDNA entre los nucleótidos 3162 y 3680, que contiene dicho gen y parte del gen NAD deshidrogenasa 1, usando los cebadores F 5'cgccttcccccgtaaatgat 3' y R 5'gccgatcagggcgtagtttg 3'. El producto de amplificación esperado es de 537 pb. Las reacciones de amplificación se realizaron en 25 µL bajo las siguientes condiciones: 2,5 µL de Buffer, 10X, Gibco BRL®; 1,25 µL de cada cebador 10µM; 0,75 µL de MgCl2 50 µM; 0,5 µL de dNTPs 10 µM; 0,125 µL de Taq polimerasa, Gibco BRL®, 5 U/µL; 2 µL de DNA a la concentración de 8 mg/µL, y se completaron los 25 µL con agua ultrapura. El perfil térmico para las amplificaciones fue el siguiente: 10 min. a 94 °C, 30 ciclos (94 °C, 30 seg; 56 °C, 30 seg; y 72 °C, 30 seg) 72 °C, 10 min. El producto de esta amplificación se visualizó por electroforesis en gel de agarosa al 1% teñida con 0,5 mg/µL de bromuro de etidio (EtBr). Estos productos se purificaron con el Kit de Promega® (Wizard® PCR Preps) y se secuenciaron por medio del método de Sanger de ddNTP acoplado a PCR; las reacciones desecuencia se realizaron en 20µL asi: 2 µL del producto amplificado y purificado equivalentes a 5 ng de DNA; 6 µL de la mezcla de PCR TRR, ABI PRISM®, BigDyeTM Terminator Cycle Secuencing (que contiene los dNTPs, ddNTPs marcados, Taq Polimerasa Gold, Buffer y MgCl2), 4 µL del cebador F 0,8 µM, y se ajustó el volumen con agua ultrapura. El perfil térmico de la reacción fue el siguiente: 25 ciclos (96 °C, 10 seg; 50 °C, 50 seg; 60 °C, 4 min).
Los productos de la reacción de secuencia se resolvieron por medio de electroforesis capilar en un Analizador Genético ABI 310, que colecta los datos a través de los programas ABI Prism 310 Collection y DNA Sequencing Analysis Software de Applied Biosystem versión 3.0. Las secuencias fueron editadas y comparadas con los programas Chromas versión 1.61 (Chromas. 1.61 edn, 2000), Visual Sequence Editor, versión 1.1 (Peters K. Visual Sequence Editor. 1.1), y GeneDoc Versión 1.1.004.
Las muestras de familiares con sospechas de poseer la mutación A3243G por línea materna se tipificaron utilizando el método PCR–RFLP para identificarla directamente, teniendo en cuenta que el cambio de AG genera un sitio de restricción para la enzima Apa I. El producto de PCR (537 pb) se sometió a digestión con dicha enzima (Promega®) con las siguientes condiciones: 0,2 µL de Apa I (10 U/µL), 0.2µL de BSA, 100X, 2 µL de Buffer y 20 µL del amplificado durante 12 horas. Los productos de las digestiones se analizaron por electroforesis en agarosa al 1,5% teñida con EtBr; la generación de dos fragmentos (456 y 81 pb) indicaba la existencia de la mutación. En un Analizador de Imágenes de BioRad, con el programa Quantity One, se cuantificó por densitometría la cantidad de mtDNA mutado, usando para ello un control de DNA con concentración conocida.
RESULTADOS Y DISCUSIÓN
Resultados moleculares
Se obtuvieron 44 secuencias (34 correspondieron a los pacientes incluidos inicialmente en el estudio, 5 a familiares de pacientes en quienes se detectó la mutación, y 5 en controles), 9 de las cuales se compararon con una secuencia consenso, encontrando cinco polimorfismos (cuatro en el gen ND1 y uno en el gen tRNALeu). En el gen ND1 seencontraron dos cambios timina citosina (TC) en las posiciones 3336 y 3396, localizados en la región codificante que no interfiere con la secuencia de la proteína por situarse en el tercer nucleótido del codón; uno citosina timina (CT), en la posición 3560, que como los anteriores es una sustitución sinónima, y finalmente, otro AG en la posición 3547, que cambia isoleucina por valina en la proteína de 11 pacientes (32,35%); en el gen tRNALeu se detectó la mutación AG en la posición 3243 (Ver figura 4) en dos pacientes.
Se comprobó que el cambio AG en la posición 3547 en el gen ND1 es un polimorfismo cuando se detecta en personas sin antecedente de mitocondriopatía,51 pero podría estar en desequilibrio de ligamiento (DL) con mutaciones implicadas en citopatías mitocondriales, por lo que se utilizó un marcador mitocondrial consistente en una deleción de 9 pb entre los genes COII y tRNALys que determinan el haplogrupo B mitocondrial.52 Se encontró que 82% de los pacientes con G en la posición 3547 presentaron la deleción, así como el 23% que tenían A, lo cual indica que dicho polimorfismo no está en DL con una mutación responsable de síndrome MELAS.
Con los resultados anteriores, la investigación se centró en analizar los dos pacientes en los que se detectó la mutación A3243G. En este sentido se hizo inicialmente un estudio clínico exhaustivo de cada uno, y luego, para el paciente M45, la cuantificación del MDNA por el medio PCR–RFLP y densitometría en sangre, músculo y saliva; en el paciente M43 la mutación solo se pudo evaluar en sangre debido a que murió recién iniciado el estudio; por último, se extendieron las genealogías de cada uno de estos pacientes con el fin de detectar la mutación en sus familiares por el método PCR–RFLP.
Análisis clínico de los pacientes
M45
Niño de diez años de edad, de sexo masculino, producto de embarazo a término, nacido por parto vaginal sin complicaciones, intervenido quirúrgicamente en las primeras horas de vida por ano imperforado. Desarrollo motor normal, con antecedentes de dislalia. A los seis años presentó cambios en el comportamiento: hablaba poco, no jugaba con otros niños, se mostraba irritable, insomne y anoréxico, y tres meses después presentó cefalea frontal seguida de inconsciencia de una hora de duración, manifestaciones que se acompañaron de fiebre y vómito; dos horas más tarde presenta un nuevo episodio caracterizado por mirada perdida, ruptura de contacto, hipotónia e inmovilidad durante una hora, seguido de estatus convulsivo focal motor del hemicuerpo izquierdo durante 72 horas, con ataxia y hemipearesia izquierda residuales por 15 días. Ocho meses después presentó nuevo estatus convulsivo focal motor, clónico izquierdo, luego del cual queda con isartria, pérdida de la memoria, inestabilidad para la marcha y dificultades de la atención y del aprendizaje. Posteriormente presentó episodios repetidos de estatus convulsivos de iguales características, que le causaron gran deterioro motor y cognitivo.
A los nueve años de edad, con peso de 20 kg, perímetro cefálico 49,4 cm y talla de 1,23 m; fue hospitalizado por crisis parciales complejas con generalización tónico clónica; al examen físico se encontró paladar ojival, implantación baja de pabellones auriculares, pectum carinatum, hipotrofia muscular generalizada, con fuerza muscular normal; no había compromiso de pares craneales, ataxia o alteraciones en la coordinación. Se trataba de un niño diestro, que no escribía los números ni las vocales, presentaba dispraxiaconstruccional y dificultades visoperceptuales; no reconocía figuras geométricas, pero sí nombraba partes del cuerpo y objetos que se le señalaban y tenía buena compresión del lenguaje. Durante la hospitalización se encontró lactacidemia de 4,4 mmol/L (valores normales 0,7–2,1mmol/L); la histología muscular, el ionograma, el pH sanguíneo y los gases arteriales fueron normales.
M43
Joven de 17 años, de sexo masculino, con historia de desarrollo psicomotor normal, y antecedentes de cefalea hemicránea izquierda pulsátil acompañada de fotopsias desde los ocho años de edad. Fue hospitalizado a los diez años por presentar un cuadro clínico de dos semanas de evolución consistente en fiebre y malestar general, dolor y debilidad muscular generalizados que le impedían la marcha. Al examen físico se encontró cuadriparesia flácida de predominio distal, arreflexia osteotendinosa generalizada, respuesta plantar flexora, marcha atáxica con steppage, signo de Gowers presente, hipotrofia muscular generalizada y sensibilidad normal; su peso fue de 20,5 kg. Un año después presentó episodio de cefalea y debilidad muscular generalizada, con recuperación completa en un mes. La concentración de proteínas en líquido cefalorraquídeo (LCR) fue de 58 mg/dL, la de glucosa de 71mg/dL y el recuento de leucocitos de 10/μL. La electromiografía mostró retardo de la latencia motora distal en ambos nervios peroneales, con respuestas de baja amplitud, sin respuesta sensitiva en surales y cubitales y con signos de denervación en músculos explorados, hallazgos indicativos de neuropatía sensitiva motora axonal.
A los 15 años se hospitaliza por cefalea global intensa de cuatro meses de evolución que no cedía fácilmente, con disminución de la agudeza visual, afasia y compromiso de la conciencia en los últimos diez días. Al examen físico presentaba frecuencia cardíaca de 80/min, frecuencia respiratoria de 20/min, estaba alerta, orientado y con disminución de la agudeza visual; el fondo de ojo fue normal. Adicionalmente se encontraron marcha atáxica y dismetría, retraso pondoestatural y del desarrollo sexual y edad ósea de 10,5–11 años. Un mes después tuvo un episodio de afasia expresiva, crisis focales motoras derechas y ceguera transitoria; se realiza una resonancia magnética (RMN) cerebral que mostró alteración deseñal en secuencia T2 con compromiso de corteza y sustancia blanca subcortical en los lóbulos temporal, occipital y parietal izquierdo, con 'efecto de masa'; en la secuencia T1 se apreciaron alteraciones de la señal de ganglios basales (globus palidum) debidas a calcificaciones. El VDRL fue 'no reactivo' la dosificación de testosterona libre fue de 0,008 pg/mL (prepuberes: 0,006–0,038 pg/mL; pubertad y adolescentes: 12,4–40 pg/mL) y la de somatomedina C de 194 ng/mL (normal: 4–100 ng/mL).
A la edad de 17 años fue hospitalizado por presentar cefalea intensa frontoparietal izquierda, parestesias y disestesias en labios, lengua y mano izquierda, crisis clónicas del miembro superior izquierdo y hemiparesia izquierda; al examen se encontró logorreico, confuso e inestable; el fondo de ojo fue normal, pero presentaba paresia del recto externo izquierdo, paresia facial central izquierda, hemiparesia izquierda, Babinski derecho, repuesta plantar neutra izquierda y hemihipoestesia izquierda. Se encontró un nivel sérico de lactato 5,9 mmol/L (normal 0.7–2.1 mmol/L), y la electromiografia fue de tipo miopático. Una tomografía axial computarizada (TAC) mostró hernia subfacial hacia la izquierda, con edema cerebral del hemisferio derecho, realce giriforme y colapso del sistema ventricular. Una resonancia magnética nuclear (RMN) cerebral mostró infarto cerebral en territorio de la arteria cerebral media derecha, con colapso ventricular ación de la línea media a la izquierda; lesión isquémica en estadio agudo. En región parietal izquierda zona hipointensa en T1 e hiperintensa en T2, con aumento del sistema ventricular ipsilateral por infarto antiguo. El EEG fue anormal, revelando enlentecimiento y desorganización de los ritmos básicos. Posteriormente presenta deterioro neurológico con cefalea severa, papiledema bilateral grado II, paresia fasciobraquial izquierda que requiere hospitalización en la unidad de cuidados intensivos, evolucionando hacia coma y muerte.
Análisis de genealogías
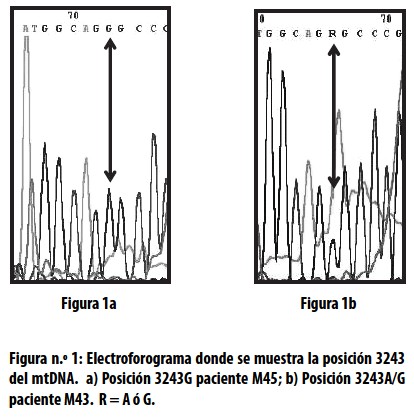
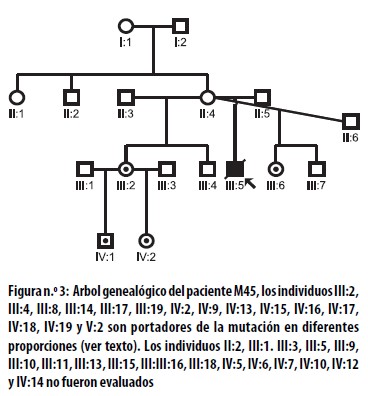
En la figura 1 se presenta la genealogía del paciente M45, en la cual se detectó la mutación en los miembros III:2, III:4, III:8, III:14, III:17, III:19, IV:2, IV:9, IV:13, IV:15, IV:16, IV:17, IV:18, IV:19 y V:2, lo cual demuestra la segregación matrilineal de dicha mutación a partir de las hijas de II:4 y II:2, a pesar de que en ellas no seencontró la mutación, debido posiblemente a que la tipificación se realizó a partir de células de la mucosa oral, en las cuales, igual que sucede en la sangre, elnivel de la mutación decrece con la edad.21,35
Al realizar la cuantificación del MDNA se encontró que el tejido muscular del paciente M45 presentaba la mayor cantidad (Ver figura 2); en los miembros de la genealogía se detectó asociación de diversos síntomas con el grado de heteroplasmia (Ver tabla 1).
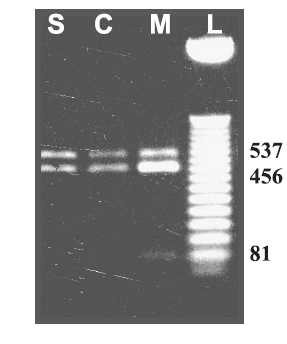
Figura n° 2: Producto de la digestión del mtDNA con la enzima ApaI, proveniente de diferentes tejidos del paciente M45. Carril C–, fragmento sin cortar; C, células de mucosa oral; M, músculo; L, marcador de peso.
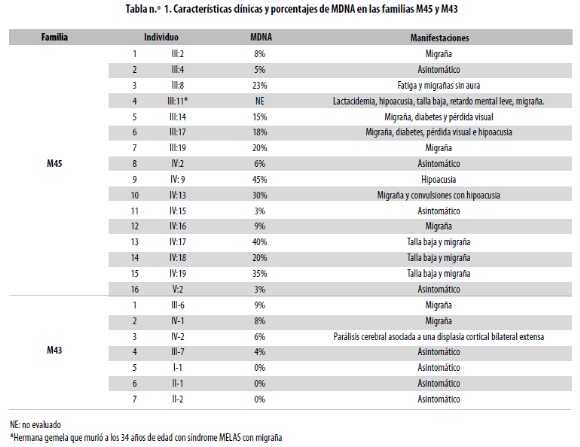
Estos resultados sugieren que según el nivel de MDNA en los distintos órganos o tejidos, la mutación A3243G puede expresarse clínicamente desde un cuadro grave de MELAS, hasta una talla baja. De los 15 individuos de la familia en los cuales se detectó la mutación, solo aquellos que tenían niveles muy bajos de MDNA, como V:2 (3% de MDNA), IV:2 (6% de MDNA), IV:15 (3% de MDNA) y III:14 (15% de MDNA), no describían ningún síntoma, incluyendo II:4 y II:2 (0% de MDNA), quienes con más 60 años eran sanas con respecto a mitocondriopatías o entidades asociadas. El mayor porcentaje (68% de MDNA) se encontró en músculo del paciente M45, diagnosticado con síndrome MELAS. Uno de los resultados más interesantes con el análisis de esta familia es que de 15 miembros que presentaron la mutación, 10 tenían migraña, lo cual sugiere que la etiología de algunas migrañas complicadas puede relacionarse con MDNA en baja proporción, y por lo tanto difícil de detectar en sangre o en células de mucosa oral.
Con respecto a la familia del paciente M43 (Figura 3 y Tabla 1) la mutación fue detectada en cuatro familiares, los cuales, como en la familia anterior, exhibían diferentes proporciones de MDNA asociados con algunos síntomas, como migraña en dos casos y parálisis cerebral espástica asociada a displasia cortical bilateral extensa en otro,. El hallazgo de migraña en dos de los cuatro individuos con la mutación en esta otra familia sugiere de nuevo alguna relación con su origen, así como con la de un umbral mínimo requerido para la aparición de síntomas.
Se ha sugerido la asociación de migraña con la mutación A3243G, considerando también la similitud entre los cuadros observados en migraña complicada y MELAS, particularmente la presencia de lesiones isquémicas occipitales asociadas a cefalea migrañosas.53
El modelo propuesto para la segregación de las mitocondrias a cada uno de los tejidos embrionarios durante el desarrollo del cigoto consistía en que se recibía de la madre un porcentaje de mtDNA mutado, el cual segregaba a las células que dan origen a los diferentes tejidos de manera estocástica, de tal manera que el efecto de las mutaciones mitocondriales depende de la cantidad de mtDNA mutado que determinados tejidos reciben durante su diferenciación, siendo más drástico en tejidos con alto gasto energético y poco poder de proliferación. Este modelo ha sido reevaluado actualmente, pues hay reportes en los cuales se demuestra que la segregación mitocondrial es determinada por genes nucleares que funcionan como QTLs.17,18 Considerando lo anterior, se decidió hacer un acercamiento al contexto genético nuclear con el fin de evaluar el efecto que podría tener sobre la cantidad de mtDNA mutado y su asociación a diferentes síntomas en los miembros de las dos familias analizadas. A este respecto, y basados en los hallazgos que se han obtenido sobre la composición genética de la población antioqueña,54 se decidió evaluar el efecto que el contexto del genoma nuclear, en lo referente a la composición ancestral, podría tener sobre el porcentaje de MDNA, y en consecuencia, en la segregación mitocondrial o en la replicación diferencial de mtDNA. Para esto se tipificaron cuatro marcadores bialélicos que discriminan las tres poblaciones ancestrales en 60 muestras tomadas a 43 pacientes y 17 familiares de los dos positivos para la mutación, y en 60 muestra tomadas a controles sanos, es decir, marcadores alelo específicos de población (PSAs); las condiciones de su genotipificación fueron tomadas de un grupo de este tipo de marcadores implementados por Parra et al.55 Los resultados que se obtuvieron fueron los siguientes: la prueba de neutralidad HWE (por sus siglas del inglés: Hardy Weinberg Equilibrium) para los 4 marcadores en las dos poblaciones de estudio no fuesignificativa en ninguna de ellas, por lo cual pueden hacerse los análisis posteriores asumiendo equilibrio; además, la medida de distancia genética entre las dos poblaciones utilizando el estadístico de Wrigth (Fst) dio como resultado un valor de 0,01 para los cuatro marcadores utilizados, lo cual significa que las diferencias que se encuentren no son debidas a estructuración entre ellas (Fst > 0,05).
Teniendo en cuenta lo anterior se espera que las diferencias en cuanto a frecuencias genotípicas sean asignadas al estatus de enfermedad entre laspoblaciones, para ello se decidió buscar dichas diferencias haciendo análisis de genotipos multilocusy utilizando como estimador el índice de ancestrí amerindia (IAA), teniendo en cuenta que las mitocondrias detectadas en los individuos con la mutación tienen origen amerindio (haplogrupo B); lo anterior se hizo tanto para los diplotipos multilocus modales de cada grupo (datos no mostrados), como para los individuos en los cuales se detecto la mutación A3243G. IAA se calcula como el logaritmo de la razón de disparidad entre la máxima verosimilitud de que un genotipo multilocus dado ocurra en la población amerindia (LA) y la que ocurra en población europea (LE), Log de LA/LE.
LA y LE se calcularon utilizando las frecuencias alélicas de las dos poblaciones y teniendo en cuenta la distribución genotípica de HWE.
El IAA para los genotipos modales fue de –0,099 y – 1,38 en casos y controles, respectivamente, lo cual quiere decir que la ancestría amerindia es mayor en el grupo de casos que en el grupo control. El cálculo delos IAA para los individuos que presentaron la mutaciónen la familia M45 se comparó con la cantidad de MDNA.Para probar si había una correlación entre MDNA e IAA,se realizó un análisis de correlación y se encontró que a mayor valor de IAA se presentaba un mayor porcentaje de MDNA, es decir, la mayor ancestría amerindia muestra una tendencia a aumentar la cantidad de MDNA, con un coeficiente de correlación de 0,8548. Este hallazgo se corrobora con el análisis de la familia del paciente M43, ya que a pesar de que no pudo hacerse análisis de correlación porque se encontraron solo dos genotipos, 19 (en M43) y 21 (en III:2, III:6, IV:1 y IV:2), se encontró que IAA presenta un valor muy alto (1,19 y 1,56, respectivamente).
Con este hallazgo puede decirse que la segregación mitocondrial en esta familia está influida por alelos de genes QTLs de origen amerindio, es decir, la cantidad de mtDNA mutado en una mitocondria de dicho origen depende de genes nucleares amerindios para su carácter heteroplásmico, y por ende para la expresiónr> patológica.
La verificación de estos hallazgos requiere la realización de más investigaciones que lo repliquen, mientras tanto, deben ser considerados con cautela.
CONCLUSIONES
– Se encuentra una fuerte asociación entre la cantidad de MDNA detectado en sangre y saliva y síntomas que se han asociado al síndrome de MELAS, como talla baja y sordera, sin embargo, lo más rescatable es la asociación clara con migraña sin aura.
– En humanos pudiera existir un efecto del contexto genético nuclear y la cantidad de MDNA, pues con nuestro trabajo se apoya la hipótesis de que la segregación mitocondrial o replicación diferencial entre MDNA y normal es dirigida por genes nucleares.
El interés por el estudio de las citopatias mitocondriales ha crecido enormemente debido al gran aumento de pacientes diagnosticados con estos trastornos, los que además se pueden expresar a cualquier edad. Igualmente, muchas de estas mutaciones se trasmiten por línea materna, como se ha indicado anteriormente, lo que hace que el diagnóstico en un individuo pueda tener implicaciones para muchas generaciones de una familia.
REFERENCIAS
1. Clay Montier LL, Deng JJ, Bai Y. Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics 2009; 36: 125–131. [ Links ]
2. Finley LW, Haigis MC. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res Rev 2009; 8: 173–188. [ Links ]
3. Devin A, Rigoulet M. Mechanisms of mitochondrial response to variations in energy demand in eukaryotic cells. Am.J.Physiol Cell Physiol 2007; 292: C52–C58. [ Links ]
4. Diaz F, Moraes CT. Mitochondrial biogenesis and turnover. Cell Calcium 2008; 44(1): 24–35. [ Links ]
5. Bertini E, D'Amico A. Mitochondrial encephalomyopathies and related syndromes: brief review. Endocr Dev 2009; 14: 38–52. [ Links ]
6. Vives–Bauza C, Gonzalo R, Manfredi G, Garcia–Arumi E, Andreu AL. Enhanced ROS production and antioxidant defenses in cybrids harbouring mutations in mtDNA. Neurosci.Lett. 2006; 391: 136–141. [ Links ]
7. van Adel BA, Tarnopolsky MA. Metabolic myopathies: update 2009. J Clin Neuromuscul Dis 2009; 10: 97–121. [ Links ]
8. Jacobs LJ, de Wert G, Geraedts JP, de Coo IF, Smeets HJ. The transmission of OXPHOS disease and methods to prevent this. Hum Reprod Update. 2006;12:119–136. [ Links ]
9. Anderson S, Bankier AT, De Bruijin MHL, Coulson AR, Drouin J. Sequence and organization of the human mitochondrial genome. Nature 1981; 290: 427–465. [ Links ]
10. Addabbo F, Montagnani M, Goligorsky MS.Mitochondria and reactive oxygen species. Hypertension 2009; 53: 885–892. [ Links ]
11. Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, et al. 2008 Strong Purifying Selection in Transmission of Mammalian Mitochondrial DNA. LoS Biol 6(1): e10. doi:10.1371/journal.pbio.0060010. [ Links ]
12. Neiman M, Taylor DR. The causes of mutation accumulation in mitochondrial genomes. Proc Biol Sci 2009; 276(1660): 1201–1209. [ Links ]
13. Shoubridge EA, Wai T. Mitochondrial DNA and the mammalian oocyte. Curr.Top.Dev.Biol. 2007; 77: 87–111. [ Links ]
14. Burnett BB, Gardner A, Boles RG. Mitochondrial inheritance in depression, dysmotility and migraine? J.Affect.Disord. 2005; 88: 109–116. [ Links ]
15. Schwartz M, Vissing J. Paternal inheritance of mitochondrial DNA. N Engl J Med. 2002 Aug 22;347:609–612. [ Links ]
16. DiMauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005; 37(3): 222–232. [ Links ]
17. Battersby BJ, Loredo–Osti JC, Shoubridge EA. Nuclear genetic control of mitochondrial DNA segregation.Nat.Genet 2003; 33: 183–186. [ Links ]
18. Farge G, Touraille S, Le Goff S, Petit N, Renoux M, Morel F, et al.. The nuclear genome is involved in heteroplasmy control in a mitochondrial mutant strain of Drosophila subobscura. Eur.J Biochem 2002; 269: 998–1005. [ Links ]
19. Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci 2008; 1142: 133–158. [ Links ]
20. Schapira AH. Mitochondrial disease. Lancet 2006; 368(9529): 70–82. [ Links ]
21. Parra MV, Carrisoza J, P,rez PF, Cornejo JW, Bedoya G, Ruiz Linares A. Genética de la citopatia micondrial MELAS (Encefalomiopatia Mitocondrial, Acidosis Lactica y Apoplejia). Medicas UIS 2006; 19: 51–55. [ Links ]
22. Finsterer J. Overview on visceral manifestations of mitochondrial disorders. Neth J Med. 2006; 64: 61–71. [ Links ]
23. Sproule DM, Dyme J, Coku J, de Vinck D, Rosenzweing E, Chung WK, et al. Pulmonary artery hypertension in a child with MELAS due to a point mutation of the mitochondrial tRNA((Leu)) gene (m.3243A > G). JIMD Short Report #096 (2007) Online. DOI 10.1007/s10545–007–0735–3. Disponible en http://www.springerlink. com/content/551762n11j2v83p3/ Consultado el 30 de junio de 2009. [ Links ]
24. Thajeb P, Dai D, Chiang MF, Shyu WC. Genotypephenotype correlation of maternally inherited disorders due to mutations in mitochondrial DNA. Taiwan J Obstet Gynecol 2006; 45: 201–207. [ Links ]
25. DiMauro S. Mitochondrial myopathies. Curr. Opin. Rheumatol. 2006; 18: 636–641. [ Links ]
26. Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC. MITOMAP: a human mitochondrial genome database–2004 update Nucleic Acids Research, 2005; 33: D611–D613 Disponible en http://nar.oxfordjournals.org/cgi/content/full/33/suppl–1/D611. Consultada en junio 20 de 2009. [ Links ]
27. Pavlakis SG, C. PP, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke–like epiosodes (MELAS):distinctive clinical syndrome. Annals of Neurology 1984; 16: 481–488. [ Links ]
28. Finnilä S, Oulu University L. Phylogenetic analysis of mitochondrial DNA: Detection of mutations in patients with occipital stroke. [archivo de internet]. Oulu Finland: University of Oulu; 2000. Disponible en http://herkules.oulu.fi/isbn9514255674/html/index.html Consultada el 30 de junio de 2009 [ Links ]
29. Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990; 348(6302): 651–653. [ Links ]
30. Matsumoto J, Saver JL, Brennan KC, Ringman JM. Mitochondrial encephalomyopathy with lactic acidosis and stroke (MELAS). Rev Neurol Dis 2005; 2: 30–34. [ Links ]
31. Gal A, Komlosi K, Maasz A, Pentelenyi K, Remenyi V, Ovary C, et al. Analysis of mtDNA A3243G mutation frequency in Hungary. Central European Journal of Medicine DOI 10.2478/s11536–009–0118–2. Disponible en http://www.springerlink.com/contentm pq81u662 346214l3/ Consultado el 30 de junio de 2009 [ Links ]
32. Salles JE, Kalinin LB, Ferreira SR, Kasamatsu T, Moises RS. Diabetes mellitus associated with the mitochondrial mutation A3243G: frequency and clinical presentation. Arq Bras Endocrinol Metabol 2007; 51: 559–565. [ Links ]
33. Valente L, Piga D, Lamantea E, Carrara, F., Uziel, G., Cudia, P et al. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim Biophys Acta 2009; 1787: 491–501. [ Links ]
34. Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat.Rev.Genet. 2005; 6: 389–402. [ Links ]
35. Procaccio V, Neckelmann N, Paquis–Flucklinger V, Bannwarth S, Jimenez R, Davila A, et al. Detection of low levels of the mitochondrial tRNALeu(UUR) 3243A>G mutation in blood derived from patients with diabetes. Mol Diagn Ther. 2006; 10: 381–389. [ Links ]
36. Cornejo JW, Parra MV, Herrera JE, Gallego JG, Pineda N, Bedoya G, et al. Análisis Molecular en 29 Pacientes con Diagnóstico Presuntivo para la Mitocondriopat¡a: Encefalopatia Mitocondrial con Acidosis Láctica y apoplejia (MELAS). Acta Neurologica Colombiana 2001; 16(3): 169–202. [ Links ]
37 Ma Y, Fang F, Yang Y, Zou L, Zhang Y, Wang Y, et al. The study of mitochondrial A3243G mutation in different samples. Mitochondrion 2009; 9: 139–143. [ Links ]
38. Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M,, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke–like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology 1992; 42: 545–550. [ Links ]
39. Rubio MA, Rinehart JJ, Krett B, Duvezin–Caubet S, Reichert AS, Soll D, et al. Mammalian mitochondria have the innate ability to import tRNAs by a mechanism distinct from protein import. Proc Natl Acad Sci 2008; 105: 9186–9191. [ Links ]
40. Kurata S, Weixlbaumer A, Ohtsuki T, Shimazaki T,Wada T, Kirino Y, et al. Modified uridines with C5–methylene substituents at the first position of the tRNA anticodon stabilize U.G wobble pairing during decoding. J Biol Chem 2008; 283: 18801–18811. [ Links ]
41. Agris PF, Vendeix FA, Graham WD. tRNA's wobble decoding of the genome: 40 years of modification. J Mol Biol 2007; 366: 1–13. [ Links ]
42. Sasarman F, Antonicka H, Shoubridge EA. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum Mol Genet 2008; 17(23): 3697–3707. [ Links ]
43. Jacobs HT, Holt IJ. The np 3243 MELAS mutation: damned if you aminoacylate, damned if you don't. Hum Mol Genet 2000; 9: 463–465. [ Links ]
44. Chomyn A, Martinuzzi A, Yoneda M, Daga A, Hurko O, Johns D, et al. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc Natl Acad Sci 1992; 89: 4221–4225. [ Links ]
45. Maassen JA, Janssen GM, t'Hart LM. Molecular mechanisms of mitochondrial diabetes (MIDD). Ann Med. 2005; 37: 213–221. [ Links ]
46. Sue CM, Lipsett LJ, Crimmins DS, Tsang CS, Boyages SC, Presgrabe CM, et al. Cochlear origin of hearing loss in MELAS syndrome. Ann Neurol. 1998; 43: 350–359. [ Links ]
47. Takeshima T, Nakashima K. MIDD and MELAS: clinical spectrum. Intern Med. 2005; 44: 276–277. [ Links ]
48. de Andrade PB, Rubi B, Frigerio F, van Den Ouweland JM, Maassen JA, Maechler P. Diabetes–associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia 2006; 49: 1816–1826. [ Links ]
49. Rozen TD, Shanske S, Otaegui D, Lu J, Young WB, Bradley K, et al. Study of mitochondrial DNA mutations in patients with migraine with prolonged aura. Headache 2004; 44: 674–677. [ Links ]
50. McEntagart M, Droogan O, Burke M, Brett F, Murphy S, Farrell M. Mitochondrial encephalopathy with lactic acidosis and stroke–like episodes (MELAS) in a Donegal kindred–clinical features and molecular genetic analysis. Ir Med J 1997; 90: 144–145. [ Links ]
51. Uusimaa J, Remes AM, Rantala, Vainionpaa L, Herna R, Vuopala K H et al. Childhood encephalopathies and myopathies: a prospective study in a defined population to assess the frequency of mitochondrial disorders. Pediatrics 2000; 105: 598–603. [ Links ]
52. Torroni A, Schurr TG, Yang CC, Szathmary EJ, Williams RC, Schanfield MS, et al. Native American mitochondrial DNA analysis indicates that the Amerind and the Nadene populations were founded by two independent migrations. Genetics 1992; 130: 153–162. [ Links ]
53. Koga Y, Akita Y, Takane N, Sato Y, Kato H. Heterogeneous presentation in A3243G mutation in the mitochondrial tRNA(Leu(UUR)) gene. Arch Dis Child 2000; 82: 407–411. [ Links ]
54. Carvajal–Carmona LG, Soto ID, Pineda N, Ortiz–Barrientos D, Duque C, McCarthy M, et al. Strong Amerind/white sex bias and a possible Sephardic contribution among the founders of a population in northwest Colombia. Am J Medical Genet 2000; 67: 1287–1295. [ Links ]
55. Parra EJ, Marcini A, Akey J, Martinson J, Balzer MA, Cooper R, et al. Estimating African American admixture proportions by use of population– specific alleles. Am J Hum Genet 1998; 63: 1839–1851. [ Links ]
Recibido: 30 de junio de 2009
Aceptado: 25 de noviembre de 2009

