










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.23 no.1 Medellín Jan./Mar. 2010
ARTÍCULO DE REVISTA
Enfermedad de Wilson: revisión del tema
Wilson's disease: a review
Yeinis Paola Espinoza Herrera1; Luis Manuel Muñoz Ruiz1; Juan Carlos Restrepo Gutiérrez2
1 Estudiante de Medicina y miembro del Grupo de Gastrohepatología, Universidad de Antioquia, Medellín, Colombia.
2 Profesor asociado, Departamento de Medicina Interna y Grupo de Gastrohepatología, Universidad de Antioquia. Hepatólogo, Hospital Pablo Tobón Uribe, Medellín, Colombia.
Correspondencia: Yeinis Paola Espinoza Herrera yeinis_eh@yahoo.com
RESUMEN
La Enfermedad de Wilson es un trastorno autosómico recesivo causado por mutaciones en el gen ATP7B que producen anormalidad en el metabolismo del cobre, con acumulación de este elemento en distintos órganos y tejidos. El diagnóstico se basa en la combinación del cuadro clínico con diversas pruebas bioquímicas, pues ninguna de ellas, aisladamente, es diagnóstica. En la actualidad se cuenta con un tratamiento efectivo para esta enfermedad, basado en la utilización de quelantes del cobre, para movilizarlo de los sitios donde se acumula y promover su excreción, así como de zinc para bloquear su absorción intestinal. El trasplante hepático es el tratamiento de elección en los pacientes con hepatopatía fulminante, así como en los que llegan a la cirrosis descompensada. En esta revisión se incluyen aspectos bioquímicos, genéticos, clínicos, diagnósticos y terapéuticos de esta enfermedad.
Palabras clave
Enfermedad de Wilson, Degeneración hepatolenticular, Metabolismo del cobre, Quelantes
SUMMARY
Wilson's disease is an autosomal recessive disorder caused by mutations in the ATP7B gene that lead to an abnormal metabolism of copper, resulting in the accumulation of this element in several organs and tissues. Its diagnosis is based on the combination of the clinical picture with various biochemical tests, neither one of which is, by itself, diagnostic of the disease. Presently there are effective treatments for EW based on the administration of chelating agents to promote mobilization of copper from the accumulation sites and its excretion. Zinc is also used in order to block the intestinal absorption of copper. Liver transplantation is the treatment of choice in patients with fulminating hepatitis, as well as in those with decompensated cirrhosis. This review includes the following aspects of Wilson's disease: biochemical, genetic, clinical, diagnostic, and therapeutic.
Key words
Chelating agents, Copper metabolism, Hepatolenticular degeneration, Wilson's disease
INTRODUCCIÓN
El neurólogo inglés Kinnear Wilson describió por primera vez en 1912 la enfermedad que lleva su nombre, como un trastorno familiar caracterizado por afectación neurológica progresiva y fatal, lesión hepática crónica y anillos de Kayser–Fleischer en la córnea.1 La enfermedad también es conocida como degeneración hepatolenticular progresiva. Su incidencia en la mayoría de las poblaciones es de uno por 30.000 nacidos vivos;2, 5 afecta por igual a hombres y mujeres, pero las manifestaciones neurológicas son más frecuentes en los primeros y la falla hepática, en las segundas.6 La EW se encuentra en todo el mundo, particularmente en los judíos del este de Europa, árabes, italianos, japoneses, chinos, hindúes y, en general, en cualquier comunidad en que sea alta la frecuencia de matrimonios consanguíneos.7 La mayor parte de los síntomas aparecen en la segunda y tercera décadas de la vida.8
La Enfermedad de Wilson (EW) es un trastorno autosómico recesivo causado por mutaciones en el gen ATP7B, localizado en el cromosoma 13 (13q14.3–q21.1),9 que codifica una ATPasa transportadora de cobre ligada a la membrana de los hepatocitos.10 La mutación genera un trastorno en la excreción de cobre en la bilis y causa acumulación de ese elemento metálico en el hígado, cerebro, riñón y córnea, dando lugar a las manifestaciones clínicas, bioquímicas e histológicas que caracterizan la enfermedad. Se han descrito alrededor de trescientas mutaciones genéticas en diferentes grupos étnicos, la más común de las cuales es la sustitución del aminoácido histidina por glutamato en la posición 1069 del gen ATP7B.11,12
En las dos últimas décadas se ha avanzado en la comprensión de la patogénesis, la biología celular y la genética molecular de la EW.8 Los adelantos en la sensibilidad de las pruebas diagnósticas han llevado a reconocer y tratar oportunamente la enfermedad.12
METABOLISMO NORMAL DEL COBRE
El cobre (Cu) es un oligoelemento esencial para muchos procesos biológicos, como cofactor importante de numerosas enzimas y determinante en la expresión de genes esenciales para la vida. Participa en el transporte de electrones, en la síntesis de colágeno y en la formación de melanina.13,14 Diariamente se necesitan 2–5 mg de este elemento que se encuentra ampliamente distribuido en los alimentos, en especial en los de origen animal: pescado, mariscos, vísceras y carnes.15,16 También está presente en leguminosas, nueces, avellanas, champiñones, granos enteros, chocolates, gelatina y productos a base de soya.17,18
El 50% del cobre ingerido se absorbe en el duodeno y el resto se excreta en las heces. El cobre se une a diversas proteínas plasmáticas como la albúmina y la transcupreína, que lo transportan hacia los tejidos, principalmente al hígado, en donde se incorpora a los hepatocitos por acción de varias cuproenzimas como la CTR1 (Copper transporter 1). En el citoplasma del hepatocito las metalotioneínas captan una parte del cobre para su almacenamiento. Proteínas denominadas chaperonas cúpricas, y específicamente la ATOX1 (Antioxidant–1), entregan el Cu a la proteína ATP7B (Transmembrane protein ATPase 7B), que lo transporta al aparato de Golgi para su incorporación a la apoceruloplasmina para formar ceruloplasmina (más del 90% del cobre plasmático es parte integrante de esta α2– glucoproteína cuyo peso molecular es 132 kD).19 El exceso de cobre se elimina por la vía biliar y se excreta principalmente en las heces, lo que evita la toxicidad del metal libre ionizado. Una cantidad menor se excreta en la orina. El proceso de excreción biliar del cobre incluye la proteína MURR1 (Mouse U1af1–rs1), que interactúa directamente con la ATP7B. (Ver Figura 1).8
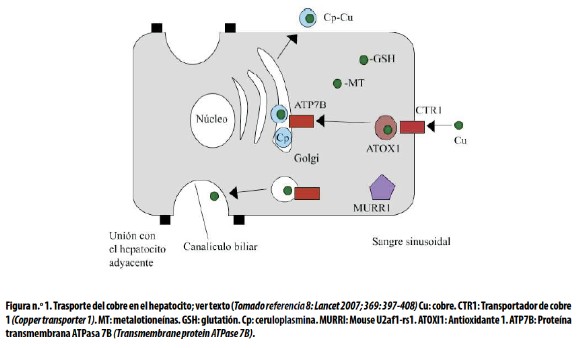
El cobre se incorpora a los hepatocitos por acción de varias cuproenzimas como la CTR1. Una vez que entra a dichas células lo capta la proteína intracitoplasmática ATOX1, la cual lo entrega a la proteína transmembrana ATP7B, encargada de trasportarlo al aparato de Golgi para su incorporación a la ceruloplasmina, forma en la cual circula la mayor parte de este elemento. Otra parte es captada por metalotioneínas para su almacenamiento. El exceso de Cu se elimina por la vía biliar (Figura 1). En la EW la deficiencia de la proteína ATP7B impide la incorporación del cobre a la ceruloplasmina dentro de los hepatocitos, lo cual da por resultado una excreción biliar alterada del elemento.3 Además, como las mutaciones en la proteína ATP7B inhiben el paso de apoceruloplasmina a ceruloplasmina, el nivel de esta última en la sangre es inferior al normal, y como esta proteína transporta la mayor parte del cobre en la sangre, en las fases iniciales de la enfermedad también está por debajo de lo normal la concentración de cobre sérico.20
No obstante, el individuo continúa la ingesta del elemento para garantizar el requerimiento diario. Es así como, al principio, el exceso de cobre se une a las metalotioneínas presentes en el hepatocito (Figura 1). Más tarde, a medida que la enfermedad avanza, se sobrepasa la capacidad almacenadora de las metalotioneínas; entonces se libera el cobre a la sangre y este elemento se acumula en otros órganos como el cerebro, los ojos y los riñones, causando las manifestaciones clínicas típicas.20, 22 En ocasiones ocurre una liberación masiva de cobre que lleva al paciente a falla hepática fulminante, hemólisis intravascular masiva y disfunción renal.22,23
CARACTERÍSTICAS CLÍNICAS
La presentación clínica de la EW es muy variable. Puede aparecer desde los 3 hasta los 70 años de edad, pero en la mayoría de los casos se manifiesta y se hace el diagnóstico antes de los 30 años, en promedio a los 17, por lo que se la considera un padecimiento de adolescentes y adultos jóvenes; hay diferentes formas de presentación, a saber: enfermedad hepática que puede ser crónica o fulminante, trastorno neurológico progresivo sin disfunción hepática evidente, hemólisis aguda o enfermedad psiquiátrica.24Las manifestaciones hepáticas son las más frecuentes (45% de los casos) y usualmente preceden a la sintomatología neurológica y psiquiátrica que aparece en 35% y 10% de los casos, respectivamente.8,16 No hay manera de predecir qué, signos y síntomas presentará un paciente dado pues el cuadro clínico depende más bien del grado de acumulación de cobre.17,25 Varias investigaciones, entre las que se encuentra la de Vélez y colaboradores, 16 le dan importancia a la identificación de mutaciones en la ATP7B para el diagnóstico preclínico de la enfermedad. Estos autores informaron sobre una nueva mutación en la ATP7B (T1232P) en pacientes de Antioquia, Colombia, relacionada con las formas neurológica y psiquiátrica de la enfermedad.
Se debe plantear la posibilidad de EW como uno de los diagnósticos diferenciales en personas entre 3 y 40 años en los siguientes casos: enfermedades hepática, neurológica o psiquiátrica de etiología desconocida; enfermedad hepática aguda con hemólisis y Coombs negativo; antecedente familiar de EW, anillo de Kayser– Fleischer en un examen con lámpara de hendidura o nivel de ceruloplasmina por debajo de 20 mg/dL; también en cualquier paciente con elevación persistente de las aminotransferasas.sup<26, 28
Presentación hepática
Las manifestaciones hepáticas de la EW son las siguientes: elevación asintomática de las aminotransferasas, hepatitis aguda o crónica, a menudo recurrente, con o sin ictericia, cirrosis en la mayoría de los casos y, ocasionalmente, falla hepática.5,10,15 Los hallazgos dependen de lo grave que sea la enfermedad y del momento en que se haga el diagnóstico.29 La presentación hepática es más común en niños que en adultos.30 y suele manifestarse entre los 8–18 años.13
El hígado es el primer órgano en que se acumula el cobre porque es el sitio primario del defecto genético.18 Los cambios histológicos iniciales incluyen el depósito de glucógeno en el núcleo de los hepatocitos y la infiltración grasa periportal. Luego hay progreso a la fibrosis y, finalmente, a la cirrosis hepatocelular en pacientes que no reciben el tratamiento oportuno.29 A menudo se presenta un cuadro clínico autolimitado semejante al de la hepatitis aguda, con malestar general, anorexia, náuseas, ictericia, niveles séricos elevados de aminotransferasas y resultados anormales de pruebas de coagulación. Algunos pacientes tienen antecedentes de ictericia autolimitada.15,30 La EW también puede presentarse como una enfermedad hepática crónica caracterizada por hepatomegalia, ascitis, esplenomegalia, nivel sérico bajo de albúmina y pruebas de coagulación con resultados anormales.31 Sin el adecuado tratamiento los pacientes pueden progresar rápidamente a la cirrosis o a la insuficiencia hepática.13
También se puede iniciar la EW como un cuadro clínico indistinguible de una hepatitis autoinmune, con fatiga, malestar general, artropatías y niveles séricos elevados de aminotransferasas e IgG y positividad para anticuerpos inespecíficos como el antinuclear y el antimúsculo liso.23 Se deben hacer estudios conducentes a un diagnóstico específico porque los tratamientos de ambas afecciones son diferentes.
Esta enfermedad también puede cursar con falla hepática que suele acompañarse de hemólisis intravascular e insuficiencia renal, debido a las grandes cantidades de cobre que se liberan hacia el torrente sanguíneo. Como típicamente no se sospecha que el paciente sea portador de EW se suele diagnosticar una hepatitis viral fulminante.32 La descompensación hepática lleva a elevación del nivel sérico de bilirrubina indirecta, reducción de la albúmina sérica y de los factores de la coagulación, ascitis, edema periférico y encefalopatía hepática. La EW ocasiona entre 6 y 12% de las fallas hepáticas fulminantes que requieren trasplante hepático de emergencia.33
Presentación neurológica
La sintomatología neurológica en la EW aparece generalmente durante la segunda y tercera décadas de la vida, pero se la ha descrito en niños de 6 a 10 años.32La ocasiona el depósito de cobre en el núcleo lenticular, el cerebelo y la sustancia negra. La afectación neurológica sigue dos patrones: trastornos del movimiento y distonía rígida.16–23 Las principales manifestaciones neurológicas encontradas en una evaluación de 119 pacientes con EW fueron: disartria (91%), alteración de la marcha (75%), risa sardónica (72%), distonía (66%), rigidez (66%), temblor (60%) y disfagia (50%). Más raramente se hallaron convulsiones y signos piramidales.34 También se pueden presentar alteraciones autonómicas, pérdida de la memoria y cefalea.
Presentación psiquiátrica
Cerca de la tercera parte de los pacientes con EW presentan algún tipo de anormalidad psiquiátrica, y alrededor del 50% han tenido trastornos de la conducta en los cinco años previos al diagnóstico, con predominio de las manifestaciones neurológicas.14 Los síntomas psiquiátricos son variados e incluyen bajo rendimiento escolar o laboral, depresión, labilidad emocional, pérdida de la inhibición sexual y psicosis.19 Se describen más comúnmente cambios en la personalidad y el comportamiento como irritabilidad y agresividad.35
Signos oculares
El anillo clásico de Kayser–Fleischer que se observa en la periferia de la córnea se debe al depósito de cobre en la membrana de Descemet.15,36 Es necesario examinar con lámpara de hendidura; también se puede observar almacenamiento de cobre en el cristalino; son las llamadas 'cataratas en girasol', que no interfieren con la visión y que, como ocurre con el anillo de Kayser– Fleischer, desaparecen con el tratamiento.23 Aunque los anillos de Kayser–Fleischer no son específicos de la EW, ya que pueden aparecer en otras hepatopatías colestásicas crónicas,15, 23 se observan en el 95% de los pacientes con las formas neurológica y psiquiátrica de la enfermedad. Pueden estar ausentes en 15–50% de los pacientes con afectación exclusivamente hepática.8,10 y están presentes en el 60% de los pacientes adultos con EW.23
Otras manifestaciones clínicas
La EW se puede asociar con varios trastornos extrahepáticos: hipercalciuria, nefrolitiasis, coledocolitiasis, artritis, osteoporosis y osteocondritis disecante. El depósito de cobre en el corazón puede desencadenar una miocardiopatía o arritmias. En las mujeres la EW puede inducir abortos espontáneos repetidos y amenorrea.37
DIAGNÓSTICO
Por su sintomatología muy inespecífica, suele ser difícil diagnosticar la EW,21,28 excepto en pacientes con la combinación clásica de enfermedad hepática crónica, temblor o distonía y anillos de Kayser–Fleischer. Sin embargo, no es habitual hallar todas las manifestaciones clínicas en un individuo.
El cuadro clínico sugestivo es la base para el diagnóstico y los hallazgos bioquímicos pueden permitir la confirmación,38,39 pero es importante reconocer que no hay una prueba diagnóstica única.3 La principal prueba de laboratorio para el diagnóstico de la EW es la medición de la ceruloplasmina en el suero, que en más del 95% de los casos está por debajo del nivel normal.40 La concentración sérica normal de esta sustancia en adultos es de 200–300 mg/dL. Si el resultado es anormal conviene hacer una medición del cobre en la orina de 24 horas, que generalmente se halla por encima de 100 µg/día. Las aminotransferasas pueden estar normales o ligeramente elevadas.31 El nivel sérico de alanina–aminotransferasa (ALT) puede ser mucho menor que el de aspartatoaminotransferasa (AST) como reflejo de la lesión de las mitocondrias hepatocíticas. La fostatasa alcalina por lo general se halla normal o baja para la edad.41
En la mayor parte de los pacientes con EW es baja la concentración sérica de cobre. El diagnóstico definitivo se hace por biopsia hepática con medición del cobre en los hepatocitos. Una concentración hepática de cobre mayor de 250 µg/g de peso seco se considera diagnóstica de la enfermedad.39Así, aunque es difícil establecer criterios diagnósticos mínimos para la EW aplicables a todos los pacientes, la enfermedad se confirma cuando se encuentra la ceruloplasmina sérica por debajo de 200 mg/dL, junto con la presencia del anillo de Kayser– Fleischer en la córnea,22 o con una concentración de cobre por encima de 250 µg/g en la biopsia de hígado.
El contenido hepático de cobre se encuentra aumentado en el 82% de los pacientes con EW, por lo que la biopsia hepática con cuantificación del cobre es el recurso diagnóstico por excelencia. Las imágenes cerebrales contribuyen muy poco al diagnóstico de la EW. La resonancia magnética se usa únicamente con el fin de identificar el tamaño de los cambios en el sistema nervioso central.10,33 En la tabla 1 se presentan los hallazgos bioquímicos característicos de la enfermedad.
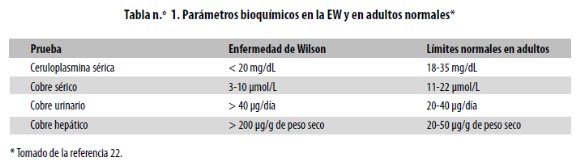
La concentración de cobre sérico libre se calcula restando el que está unido a ceruloplasmina del total en el suero.42 Este parámetro no es muy útil en el diagnóstico pero sí lo es para vigilar la respuesta al tratamiento.
TRATAMIENTO
Los tratamientos para la EW se han centrado en el empleo de agentes quelantes del cobre para movilizarlo de los sitios donde se acumula y promover su excreción; todos ellos tienen efectos secundarios importantes. Se los puede administrar acompañados o no por zinc, dependiendo del grado de descompensación hepática. La terapia farmacológica en la EW se debe mantener de por vida, y la elección del medicamento depende más de la opinión del médico tratante que de los datos comparativos disponibles.43
La penicilamina, químicamente relacionada con la cisteína, introducida en 1956 por Walshe, es eficaz en la mayoría de los pacientes con EW;32 contiene grupos sulfhidrilo que aumentan la excreción urinaria de cobre, y se administra por vía oral a una dosis inicial de 250–500 mg/día, con incrementos de 250 mg cada cuatro a siete días, hasta llegar a una dosis de mantenimiento de 750–1.000 mg/día dividida en 2–4 tomas. Es el medicamento más eficaz para eliminar el cobre, pero produce muchos efectos adversos en el 74% de los casos, y su toxicidad puede empeorar las manifestaciones neurológicas en la mitad de los pacientes.3,11 Por eso, aunque por muchos años se la consideró como el mejor tratamiento para los pacientes con EW, actualmente está siendo reemplazada por fármacos como la trientina, cuyos efectos secundarios son menores. Algunos de los efectos adversos de la penicilamina son: interferencia en la formación de elastina y colágeno, leucopenia, trombocitopenia, lupus eritematoso sistémico, nefritis, pénfigo, ulceraciones orales y miastenia gravis. Estos efectos colaterales graves requieren la suspensión inmediata del fármaco y el uso de un quelante distinto.44
La trientina, introducida también por Walshe, es el tratamiento de segunda línea en pacientes que no toleran la penicilamina; su mecanismo de acción es similar al de esta última, aumenta la excreción urinaria de cobre y puede interferir con su absorción intestinal. Otra alternativa es utilizar sulfato de zinc por vía oral para promover la excreción de cobre en las heces, puesto que interviene en la absorción intestinal del cobre por los enterocitos y estimula la síntesis hepática de metalotioneína.45
En pacientes con alteraciones hepáticas tales como hepatitis o cirrosis sin signos mayores de descompensación y en ausencia de sintomatología neurológica, el zinc es el tratamiento de elección, aunque algunos autores aconsejan acompañarlo siempre con un quelante. En presencia de signos claros de descompensación hepática, el tratamiento adecuado es un quelante (preferiblemente trientina) más zinc, pero hay que tener en cuenta que estos fármacos deben administrarse con un intervalo mínimo de una hora para evitar interacciones que disminuyan sus efectos terapéuticos. El tetratiomolibdato de amonio se usa como tratamiento de la EW neurológica grave. Este fármaco interfiere con la absorción de cobre en el intestino y se une con alta afinidad al cobre plasmático.32
Debido a la toxicidad de los medicamentos anticúpricos, es de gran importancia ejercer una adecuada vigilancia del paciente con hemoleucograma y uroanálisis; la frecuencia con que se hacen estos exámenes es como sigue: cada semana en el primer mes de tratamiento; cada dos semanas en los meses dos y tres; mensualmente en los meses cuatro y cinco y semestralmente.de ahí en adelante.46 Con el fin de analizar los resultados luego de instaurado el medicamento se miden el cobre sérico libre y la excreción urinaria de cobre en 24 horas. El tratamiento con zinc no requiere vigilancia estricta pues sus efectos indeseados no son muy importantes y se manifiestan tan sólo en el 10% de quienes lo utilizan.
Dieta
Como parte fundamental del tratamiento, los pacientes con EW deben mantener de por vida una dieta baja en cobre: menos de 1 mg por día.47
PRONÓSTICO Y ANÁLISIS DE LA MUTACIÓN
El estudio genético es costoso y difícil por el gran número de mutaciones en el gen implicado (alrededor de 300);41 por ello no es muy útil para el diagnóstico.45,48 Teniendo en cuenta que la EW es un trastorno autosómico recesivo, y que la mejor evolución se asocia con el tratamiento iniciado en el período presintomático, es necesario detectar los marcadores genéticos de la enfermedad en todos los familiares del paciente en primer grado. Se acepta que los pacientes con EW tienen buen pronóstico si la enfermedad se diagnostica a tiempo y se trata adecuadamente, pero cuando hay cirrosis la única alternativa de curación es el trasplante hepático.49
TRASPLANTE HEPÁTICO
No tratada, la EW es progresiva y fatal. Por ello el trasplante hepático es el tratamiento de elección en quienes la sufren y tienen falla hepática fulminante; también cuando la enfermedad cursa con cirrosis descompensada o el paciente no responde a la terapia farmacológica.27,50,51 Este procedimiento corrige el defecto bioquímico de base y alarga la supervivencia. Así lo comprobaron Sevmis y colaboradores,27 quienes hicieron seguimiento a 24 pacientes con EW sometidos a trasplante de hígado entre septiembre de 2001 y junio de 2007, observando que en todos los pacientes el nivel de ceruloplasmina volvía al rango normal meses después del trasplante y que disminuían la excreción urinaria de cobre y las manifestaciones neurológicas.27 El pronóstico de supervivencia tras un trasplante es mejor en los pacientes con enfermedad hepática crónica avanzada (90%) que en aquellos con falla hepática fulminante (73%).49,52
CONCLUSIÓN
La EW es un trastorno autosómico recesivo del metabolismo del cobre, que produce diversas manifestaciones clínicas. Afortunadamente su frecuencia es baja en la población general. No obstante, su diagnóstico y tratamiento le plantean un reto al clínico, quien debe hacer una historia clínica completa y sospechar la enfermedad en toda persona, sin importar la edad, con enfermedad hepática o neuropsiquiátrica de origen desconocido y, también, cuando observe de manera casual el anillo de Kayser Fleischer. El diagnóstico de la EW es el resultado de la interpretación de pruebas clínicas, bioquímicas e histológicas. Se inicia con la cuantificación del cobre en la orina de 24 horas, para luego determinar los niveles séricos de cobre y ceruloplasmina. La prueba diagnóstica definitiva es la biopsia hepática percutánea con medición del cobre. Las pruebas imaginológicas no son diagnósticas de la enfermedad, pues las imágenes son en extremo inespecíficas y dependen de la fase en que se encuentre la enfermedad. El análisis de la mutación para propósitos investigativos es un procedimiento que está adquiriendo importancia en la caracterización demográfica de la enfermedad. Una vez diagnosticada la EW se debe iniciar la terapia farmacológica, que es efectiva en el 95% de los casos. Se recomienda estudiar a los familiares del caso índice para hacer diagnósticos tempranos y tratamientos oportunos.
REFERENCIAS BIBLIOGRÁFICAS
1. Walshe JM. History of Wilson's disease: 1912–2000. Mov disord 2006; 21: 142–147. [ Links ]
2. Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli–Vergani G. Wilson's disease in children: 37–year experience and revised Kin's score for liver transplantation. Liver Transpl 2005; 11: 441–448. [ Links ]
3. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long–term outcome of Wilson's disease: a cohort study. Gut 2007; 56: 115–120. [ Links ]
4. Petrasek J, Jirsa M, Sperl J, Kozak L, Taimr P, Spicak J, et al. Revised King's College score for liver transplantation in adult patients with Wilson's disease. Liver Transpl 2007; 13: 55–61. [ Links ]
5. Gheorghe L, Popescu I, Iacob S, Gheorghe C, Valdan R, Costantinescu A, et al. Wilson's disease: A challenge of diagnosis. The 5–year experience of a tertiary centre. Rom J Gastroenterol 2004; 13: 179–185. [ Links ]
6. Magalini SI, Magalini SC. Wilson (S.A.K). Dictionary of medical syndromes. 4a ed. Philadelphia: Lippincott– Raven; 1997, 1042 p. [ Links ]
7. Sherlock S, Dooley J. Diseases of the Liver and Biliary System. 11a ed. Oxford: Blackwell Science; 2002, 706 p. [ Links ]
8. Ala A, Walker A, Ashkan K, Dooley J, Schilsky M. Wilson's disease. Lancet 2007; 369: 397–408. [ Links ]
9. Vélez C, Jiménez M, Moreno S, Ramírez L, Correa G, Lopera F. New mutation (T1232P) of the ATP–7B gene associated with neurologic and neuropsychiatric dominance onset of Wilson's disease in three unrelated Colombian kindred. Neuroscience Letters 2004; 37: 360–364. [ Links ]
10. Ferenci P. Pathophysiology and clinical features of Wilson disease. Metab Brain Dis 2004; 19: 229–239. [ Links ]
11. Firneisz G, Lakatos PL, Szalay F, Polli C, Glant TT, Ferenci P. Common mutations of ATP7B in Wilson disease patients from Hungary. Am J Med Genet 2002; 108: 23–28. [ Links ]
12. Auth MK, Kim HS, Beste M, Bonzel KE, Baumann U, Ballauf A, et al. Removal of metabolites, citokines and hepatic growth factors by extracorporeal liver support in children. J Pediatr Gastroenterol Nutr 2002; 40: 54–59. [ Links ]
13. Ferenci P. Review article: diagnosis and current therapy of Wilson's disease. Aliment Pharmacol Ther 2004; 19: 157–165. [ Links ]
14. Sarkar B. Copper transport and its defect in Wilson disease: Characterization of the copper–binding domain of Wilson disease ATPase. J Inorg Biochem 2000; 79: 187–191. [ Links ]
15. Roberts E, Schilsky M. Diagnosis and treatment of Wilson disease: An update. Hepatology 2008; 47: 2089–2111. [ Links ]
16. Vélez C, Jiménez M, Lopera F. Enfermedad de Wilson. Medicina & Laboratorio 2002; 10: 59–67. [ Links ]
17. Ferenci P, Caca K, Loudianos G, Mieli–Vergani G, Tanner S, Sternlieb I. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003; 23: 139–142. [ Links ]
18. Gitlin N. Wilson's disease: The scourge of copper. J Hepatol 1998; 28: 734–739. [ Links ]
19. Klomp A, Tops B, Van den Beerg I, Beerger R, Klomp L. Biochemical characterization and subcellular localization of human copper transporter 1 (hCTR1). Biochem J 2002; 36: 497–505. [ Links ]
20. Ferenci P, Steindl–Munda P, Vogel W, Jessner W, Gschwantler M, Stauber R. Diagnostic value of quantitative hepatic copper determination in patients with Wilson's disease. Clin Gastroenterol Hepatol 2005; 3: 811–818. [ Links ]
21. Medici V, Trevisan CP, D'Inca R, Barollo M, Zancan L, Fagiuoli S. Diagnosis and management of Wilson's disease: results of a single center experience. J Clin Gastroenterol 2006; 40: 936–941. [ Links ]
22. Gow P, Smallwood R, Angus P, Smith A, Wall A, Sewell R. Diagnosis of Wilson's disease: an experience over three decades. Gut 2000; 46: 415–419. [ Links ]
23. Roberts E. Wilson' disease. Medicine 2006; 35: 93–95. 24. Kumagi T, Horiike N, Abe M, Kurose K, Luchi H, Masumoto T, et al. Small hepatocellular carcinoma associated with Wilson's disease. Intern Med 2005; 44: 439–443. [ Links ] [ Links ]
25. Riordan S, Williams R. The Wilson's disease gene and phenotypic diversity. J Hepatol 2001; 34: 165–171. [ Links ]
26. Zakim, D. Boyler, T. Hepatology: A textbook of liver disease. 4a ed. Philadelphia: Saunders; 2003. 1568 p. [ Links ]
27. Sevmis H, Karakayali I, Aliosmanoglu U, Yilmaz F, Ozcay A, Torgay G. Liver transplantation for Wilson's disease. Transplant Proc 2008; 40: 228–230. [ Links ]
28. Shiono Y, Hayashi H, Wakusawa S, Yano M. Ultrastructural identification of iron and copper accumulation in the liver of a male patient with Wilson disease. Med Electron Microsc 2001; 34: 54–60. [ Links ]
29. Akpinar E, Akhan O. Liver imaging findings of Wilson's disease. Eur J Radiol 2007; 61: 25–32. [ Links ]
30. Cox DW, Roberts EA. Enfermedad de Wilson. En: Sleinsenger M, Fordtran J, Feldman M, Lawrence S. Enfermedades gastrointestinales y hepáticas: Fisiopatología, diagnóstico y tratamiento. 7ª ed. Argentina: Mé,dica Panamericana; 2004: 1344–1353. [ Links ]
31. Roberts E, Schilsky M. A Practice Guideline on Wilson Disease. Hepatology 2003; 37: 1475–1492. [ Links ]
32. Restrepo JC. Enfermedades metabólicas del hígado. En: Fundamentos de Medicina: Gastroenterología y Hepatología, 5a ed. Medellín (Col): CIB; 2004. 221–225. [ Links ]
33. Shiono Y, Wakusawa S, Hayashi H, Takikawa T, Yano M, Okada T. Iron accumulation in the liver of male patients with Wilson disease. Am J Gastroenterol 2001; 96: 3147–3151. [ Links ]
34. Restrepo D, Calle J. Aspectos neuropsiquiátricos de la enfermedad de Wilson y la esclerosis múltiple. Rev Colomb Psiquiat 2007; 36 (Suppl.1): 126–138. [ Links ]
35. Mercer J. The molecular basis of copper–transport diseases. Trends Mol Med 2001; 7: 64–69. [ Links ]
36. Koppikar S, Dhawan A. Evaluation of the scoring system for the diagnosis of Wilson's disease in children. Liver Int 2005; 25: 680–681. [ Links ]
37. Walshe J, Waldenstrom E, Sams V, Nordlinder H, Westermark K. Abdominal malignancies in patients with Wilson's disease. QJM 2003; 96: 657–662. [ Links ]
38. Loudianos G, Gitlin JD. Wilson's disease. Semin Liver Dis 2000; 20: 353–364. [ Links ]
39. Brewer G. Recognition, diagnosis, and management of Wilson's disease. Proc Soc Exp Biol Med 2000; 223: 39–46. [ Links ]
40. Tao Y, Gitlin J. Hepatic copper metabolism: Insights from genetics disease. Hepatology 2003; 37: 1241–1247. [ Links ]
41. Mak CM, Tam S, Fan ST, Liu CL, Lam CW. Wilson's disease: A patient undiagnosed for 18 years. Hong Kong Med J 2006; 12: 154–158. [ Links ]
42. Gupta A, Aikath D, Neogi R. Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype–phenotype correlation in patients. Hum Genet 2005; 118: 49–57. [ Links ]
43. Van Wassenaer–van Hall H, Van den Heuvel A, Algra A, Hoogenraad TU, Mali W. Wilson disease: Findings at MR imaging and CT of the brain with clinical correlation. Radiology 1996; 198: 531–536. [ Links ]
44. Askari FK, Greenson J, Dick RD. Treatment of Wilson's disease with zinc. Initial treatment of the hepatic decompensation presentation with trientine and zinc. J Lab Clin Med 2003; 142: 385–390. [ Links ]
45. Brewer G, Askari F. Wilson's disease: clinical management and therapy. J Hepatol 2005; 42:S13–S21. [ Links ]
46. Brewer G. Wilson's disease: A clinician's guide to recognition, 1a ed. Boston: Kluwer Academic; 2001, 158 pages. [ Links ]
47. Bressman SB, Moadelli. MD virtual University (MDVU). (2004, Abril 13). Wilson disease. Disponible en www.mdvu.org/library/disease/wd/wd_dia.html. Consultado abril 16 de 2008. [ Links ]
48. Schilsky M. Wilson's disease: Genetic basis of copper toxicity and natural history. Semin Liver Dis 1996: 16: 83–95. [ Links ]
49. Emre S, Atillasoy E, Ozdemir S, Schilsky M, Rathna C, Thung S. Orthotopic liver transplantation for Wilson's disease: A single–center experience. Transplantation 2001; 72: 1232–1236. [ Links ]
50. Haberal M, Moray G, Karakayali H, Arslan G, Boyacioglu S, Baysal C, et al. Liver transplantation for Wilson's cirrhosis: one center's experience. Transplant Proc 1999; 31: 3160–3161. [ Links ]
51. Bellary S, Hassanein T, Van Thiel DH. Liver transplantation for Wilson's disease. Acta Neurol Scand 1995; 92: 405–408. [ Links ]
52. Steindl P, Ferenci P, Dienes H. Wilson's disease in patients presenting with liver disease. A diagnostic challenge. Gastroenterology 1997; 113: 212–218. [ Links ]
Recibido: febrero 06 de 2009
Aceptado: marzo 04 de 2009

