










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.23 no.3 Medellín July/Sept. 2010
PRESENTACIÓN DE CASOS
Aspectos imaginológicos del sarcoma granulocítico en la cara y el cráneo de niños y jóvenes: informe de cinco casos y revisión de la literatura
Radiological findings of granulocytic sarcoma in the face and skull of children and young persons: report of five cases and literature review
Elcy Medina Medina1; Rafael Cárdenas Restrepo2 ; Beatriz Molinares Arévalo2; Daniel Arias Betancur3;Ana María Valencia Cadavid3; José William Cornejo Ochoa4
1 Profesora asociada de Neurorradiología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. elcy.medina@gmail.com
2 Residente de Radiología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
3 Estudiante de pregrado, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
4 Profesor titular de Neurología-Epidemiologia, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
RESUMEN
Introducción: el sarcoma granulocítico (SG) es una neoplasia maligna cuya incidencia es de 2,9% a3,1% en pacientes con leucemia mieloide (LM) o enfermedades mieloproliferativas. Se presentahabitualmente en hombres y en población africana, asiática y suramericana.
Objetivo: describir las características imaginológicas del SG en la cara y el cráneo de cinco niños yjóvenes.
Pacientes y métodos: presentamos cinco pacientes con SG en la cara y la base del cráneo. A cuatrode ellos se les hizo tomografía computarizada (TC) y a tres, resonancia magnética (RM) cerebral.Cuatro presentaron una masa de tejido blando en la órbita, dos tenían afectación ósea y otro revelólesión en el sistema nervioso central. En cuatro se diagnosticó leucemia mieloide aguda (LMA) .
Conclusión: el SG puede manifestarse con invasión orbitaria y craneofacial en niños y adultosjóvenes. Usualmente los pacientes consultan por proptosis y edema orbitario. Con este cuadroclínico el SG es la primera probabilidad diagnóstica en el contexto de la LMA o las mielodisplasias. Enotras situaciones clínicas se debe hacer diagnóstico diferencial con complicaciones de sinusitis,rabdomiosarcoma, linfoma de la órbita y otras neoplasias. La imaginología demuestra invasión detejidos blandos e infiltración ósea. Es muy característico del SG afectar en un comienzo la paredlateral o la superior de la órbita. En algunos casos simula abscesos. El diagnóstico se confirma porhistopatología.
Palabras clave
Cloroma, Enfermedades mieloproliferativas, Leucemia mieloide, Neoplasia mieloide, Sarcomagranulocítico craneofacial
SUMMARY
Introduction: Granulocytic sarcoma (GS) is a rare malignant neoplasia, with an incidence rate of 2.9% to 3.1% in patients with myeloid leukemia or myeloproliferative diseases. Usually it affects males, of African, Asian and South American populations.
Objetive: To describe the radiological characteristics of GS in the face and skull of children and young people.
Patients and methods: We report five patients with GS in the face and skull. Computerized tomography (CT) was carried out in four and orbital and brain magnetic resonance (MRI) in three. Out of the five, four had soft tissue masses in the orbit, two had bone infiltration, and in one there was a central nervous system lesion. In four patients acute myeloid leukemia was demonstrated.
Conclusion: GS may affect the orbit, the face and the skull of children and young adults; it manifests with exophthalmia and orbital edema. It must be considered as a diagnostic possibility in the context of myeloid leukemia and myelodysplasias. Otherwise, differential diagnosis should be made with complications of sinusitis, orbital rhabdomyosarcoma, orbital lymphoma and other tumors. Imaging studies may help in the diagnosis by revealing soft tissue masses and areas of bone infiltration. In some cases GS may resemble abscesses. Diagnosis must be confirmed by histopathology.
Key words
Chloroma, Craneofacial granulocytic sarcoma, Myeloid leukemia, Myeloid neoplasia, Myeloproliferative diseases
INTRODUCCIÓN
Allen Burns en 1811 fue el primero en describir el sarcoma granulocítico (SG) 1 o cloroma. Se trata de una neoplasia maligna rara, cuya tasa de incidencia es de 2,9 a 3,1% en pacientes con leucemia mieloide (LM) o trastornos mieloproliferativos. El término cloroma se acuñó debido a su coloración verdosa, producida por la mieloperoxidasa del tejido mieloide inmaduro al contacto con el aire. Rappaport sugirió el término SG para incluir los tumores que no fueran verdes.2 Puede presentarse en asociación con la leucemia mieloide aguda (LMA) , con trastornos mielodisplásicos con transformación leucémica o leucemia mieloide crónica con crisis blástica inminente, y en pacientes sin evidencia de LMA ni de trastorno mieloproliferativo o síndrome mielodisplásico.3
La gran mayoría de los pacientes no leucémicos con SG desarrollan LMA varios meses después. El tiempo promedio entre el diagnóstico del SG y el comienzo de la leucemia es de 10,5 meses. Sin embargo, se han informado pacientes libres de enfermedad durante 3,5 a 16 años después de la presentación inicial del SG.3 Se manifiesta habitualmente en los subtipos M4 (mielomonocítico) y M5 (monocítico) de la LMA. Usualmente se presenta en hombres entre los 6 y 77 años y en población africana, asiática, sudamericana y de descendencia española.1
Para algunos autores el SG se origina en la médula ósea y alcanza el subperiostio por los canales de Havers.4 Puede afectar el cráneo y la cara, el esternón, las costillas y las regiones proximales de los huesos largos; además, tejidos blandos, piel y ganglios. En algunas ocasiones puede afectar estructuras perineurales y epidurales y así causar compresión del sistema nervioso central y del periférico.5,6 La edad media de aparición es de 8 años. Entre los síntomas generales del SG se pueden encontrar dolor localizado, generalizado o radicular, déficit motor, hipoestesia y trastornos esfinterianos. Son infrecuentes las manifestaciones extraóseas, entre ellas las debidas al compromiso de ovarios, mamas, pulmones, intestinos, riñones e hígado. La invasión orbitaria se presenta generalmente en niños y adultos jóvenes, Cuando se diagnostica primero la leucemia mieloide sistémica, las manifestaciones orbitarias ocurren hasta 94 meses después del diagnóstico; son ellas: proptosis, edema orbitario, dacrioadenitis, afectación palpebral, conjuntival, del iris y de la coroides.
El diagnóstico de este tumor se puede sugerir mediante tomografía computarizada (TC) , resonancia magnética (RM) , mielo-TC o mielo-RM. La confirmación se hace por biopsia a cielo abierto o guiada por tomografía. La apariencia radiológica es de masas isodensas con el músculo en TC e hipointensas en T1 y T2 debido a su alta celularidad, con realce leve o moderado, algunas veces anular que puede simular un absceso. De preferencia afecta la pared lateral de la órbita con localización extraconal y finalmente invasión del canal óptico.7 Al examen macroscópico tiene configuración nodular y es de consistencia maciza. Microscópicamente se observan células indiferenciadas con núcleos hipercromáticos y nucléolos prominentes, cuyo citoplasma contiene granulaciones finas. Para una adecuada diferenciación se requieren coloraciones que evidencien la positividad para las esterasas (tinción con naftol-ASD-cloroacetato esterasa) y peroxidasas. Si el resultado de esas coloraciones es negativo, se sugiere el diagnóstico de granuloma eosinofílico.
En la microscopía electrónica se observan granulocitos inmaduros en un estroma laxo. Las células tienen gránulos elipsoidales con bastones cristalinos iguales a los bastones de Auer y los cuerpos Phi de las células leucémicas.8
Hematológicamente se puede hallar desde un infiltrado difuso de células maduras de la estirpe granulocítica hasta blastos mieloides, monocitoides o megacariocíticos. Por inmunohistoquímica e inmunofenotipo se pueden observar anomalías citogenéticas características como la traslocación 8;21, alteraciones del cromosoma 16, traslocaciones en el cromosoma 11q 23 y la trisomía del cromosoma 8.4,9
Debido a que el SG es una manifestación de la LMA, el tratamiento es el de esta última. El protocolo en Medellín es el Berlín-Frankfurt-Munster (BFM 87) modificado por el Grupo Argentino de Tratamiento de la Leucemia Aguda.
En la evaluación de la LMA lo primero que se hace es un hemograma con extendido de sangre periférica y un aspirado de médula ósea en los días 1,15 y 33 del estudio, con el objetivo de buscar alteraciones en el recuento y la morfología celulares. Se deben hacer pruebas para definir el inmunofenotipo por citometría de flujo. La LMA se clasifica de M0 a M7; para ello es de gran importancia hacer el cariotipo con hibridación in situ fluorescente (FISH) , para definir si hay alteraciones cromosómicas.
Pruebas complementarias son: ecografía abdominal y radiografía de tórax, con las que se verifica la presencia de hepatoesplenomegalia y la afectación ósea, pulmonar o mediastinal, respectivamente. Si se sospecha invasión del sistema nervioso central se debe hacer un citoquímico de líquido cefalorraquídeo. Al confirmarla, el tratamiento consiste en quimioterapia intratecal y radioterapia. Sin embargo, según algunos autores, la radioterapia en pacientes con SG no tiene ninguna influencia en atenuar o curar la enfermedad.10
El propósito del presente estudio fue revisar retrospectivamente los aspectos clínicos e imaginológicos de cinco casos de SG en la cara y el cráneo; todos habían sido evaluados por uno de los autores (EM) entre los años 1996 y 2007.
PACIENTES
Caso n.º 1
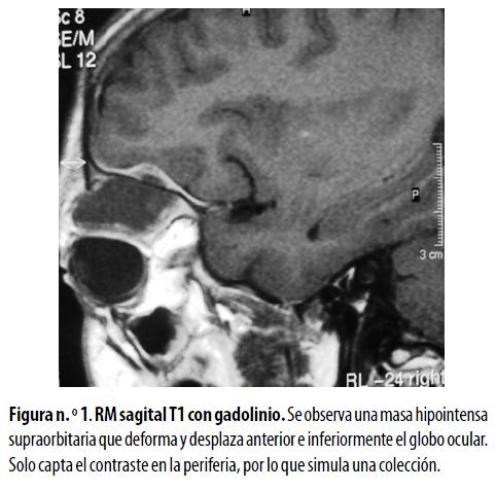
Niña de 6 años, residente en zona rural, llevada a la consulta porque presentaba proptosis progresiva derecha. No refería alteración visual ni otros síntomas de importancia. El desarrollo psicomotor era normal para la edad. Con la sospecha clínica de una masa retrocular, se le hizo una TC de órbitas que mostró una imagen con densidad de tejidos blandos localizada en el techo de la órbita con extensión al espacio retrocular; la lesión afectaba el músculo recto superior pero no invadía el globo ocular, ni el nervio óptico ni las superficies óseas. Ante la necesidad de caracterizar mejor el alcance de la lesión en los tejidos blandos se hizo una RM que reveló una lesión hipointensa con estos en las imágenes sopesadas en T1, con realce anular después de la administración de gadolinio (figura nº. 1) e hipointensidad en las secuencias ponderadas en T2 que evidenció un alto contenido celular. La biopsia demostró un sarcoma granulocítico. Como no existían antecedentes ni sospecha de leucemia, se le hicieron estudios complementarios de médula ósea y sangre periférica que evidenciaron una LMA como enfermedad de base.
Caso n.º 2
Niño de 8 meses traído a la consulta por la presencia de una masa dura, indolora en el maxilar derecho, de tres meses de evolución, que afectaba la encía. Había sido tratado con antibióticos en muchas ocasiones pero la masa seguía aumentando de tamaño y produjo deformidad facial.
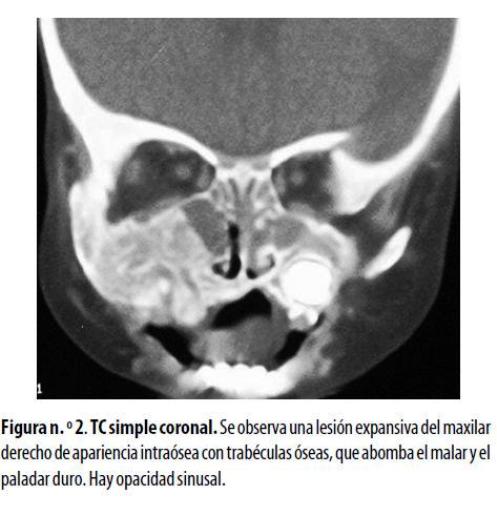
La TC de los senos paranasales mostró una lesión mixta de tejido blando con trabéculas óseas engrosadas, ligeramente expansiva, que afectaba el malar y el maxilar derechos y el paladar duro, pero sin destrucción ósea. Había contacto íntimo con las raíces dentarias pero no lesión subcutánea. Se observó engrosamiento de la mucosa de los senos maxilares y las celdillas etmoidales estaban ocupadas por material con densidad de tejido blando. Se observó también aumento bilateral de los tejidos blandos intranasales (figura n.º 2) . En una biopsia inicial del maxilar derecho se diagnosticó linfoma de Burkitt; se inició quimioterapia pero la respuesta fue mala; requirió varias hospitalizaciones por cuadros de agranulocitosis y síndromes febriles. Otro estudio histopatológico mostró múltiples espículas óseas con áreas necróticas y algunos fragmentos de tejido blando como músculo y tejido conectivo. El estudio inmunohistoquímico reveló la existencia de un SG. Se cambió el esquema de quimioterapia, lo que fue seguido de franca mejoría clínica y disminución del tamaño de la lesión.
Caso n.º 3
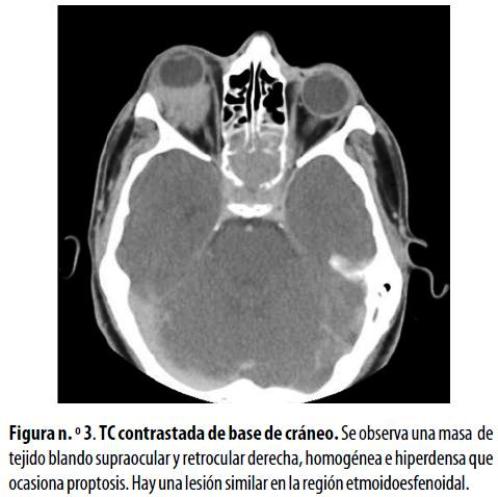
Hombre de 18 años en recaída de LMA, quien consultó por dolor orbitario y proptosis derecha. La TC mostró una masa homogénea de tejido blando supraocular y retrocular en la órbita derecha, que ocasionaba la proptosis, y lesión erosiva en la región esfenoetmoidal (figura n.º 3) . En vista de la progresión de sus síntomas visuales fue sometido a radioterapia con buena tolerancia; sin embargo, no se describieron los controles clínicos adicionales después de la terapia.
Caso n.º 4
Niño de 9 años que consultó por LMA, cloroma facial, epistaxis y afectación del VII par. La TC demostró una gran masa de tejido blando similar al músculo en la pared externa de la órbita derecha con destrucción ósea, proptosis e invasión a la fisura esfenomaxilar. Se observó también una masa similar en la región inframastoidea derecha. La RM no demostró lesión intracraneana. Tuvo recaída con exacerbación de su sintomatología; su pronóstico era malo porque no se pudo hacer trasplante de médula ósea. No regresó a la consulta.
Caso n.º 5
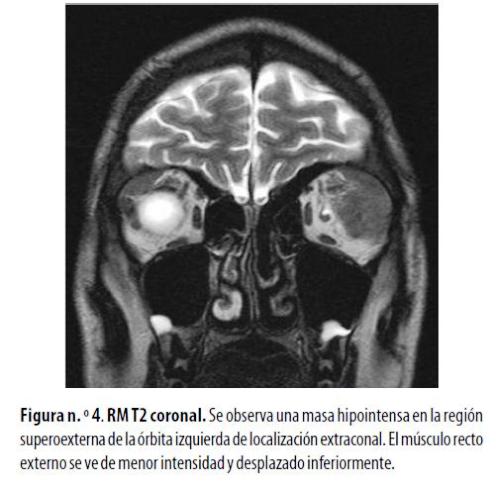
Hombre de 17 años con diagnóstico de LMA que afectaba el SNC. Presentó proptosis izquierda. En las imágenes ponderadas en T1 la RM demostró una lesión hipointensa y fusiforme en la región superoexterna de la órbita izquierda, que infiltraba el músculo recto externo. Presentó captación homogénea a la administración de gadolinio y se observó captación meníngea nodular en la región frontal. Había signos de reemplazo por grasa de la médula ósea de la calota craneana por infiltración mieloide (figura n.º 4) . Se le administraron radioterapia y quimioterapia y presentó mejoría en el primer año de tratamiento.
DISCUSIÓN
El SG o cloroma es una manifestación extramedular de la LMA o de síndromes mielodisplásicos, que puede presentarse en el mesénquima de cualquier parte del organismo. Usualmente hace parte del cuadro clínico de pacientes que ya tienen el diagnóstico de alguna de estas entidades, pero también puede ser la primera manifestación de una enfermedad de base, como ocurrió en el caso n.º 1; además, puede aparecer en el período de remisión de la leucemia. El diagnóstico suele ser muy difícil por lo que se recurre a la biopsia para estudios anatomopatológico e inmunohistoquímico,1 como en los casos n.º 1 y 2. En general el SG se presenta en niños y jóvenes con LMA,4 como ocurrió en cuatro de nuestros cinco pacientes. Por la afectación orbitaria se debe hacer diagnóstico diferencial con complicaciones de la sinusitis, seudotumor orbitario, rabdomiosarcoma, linfoma, plasmocitoma, histiocitosis, entre otras enfermedades.
La localización superoexterna en la órbita se halló en cuatro de los cinco pacientes de esta serie, acorde con lo descrito frecuentemente para el SG. Lo mismo se puede afirmar de la afectación parameníngea que presentó el paciente n.º 5. Con relación al sexo, la mayoría de las series muestran un predominio masculino. Cuatro de nuestros cinco pacientes eran hombres, todos ellos nacidos y residentes en Colombia, lo que se correlaciona con la alta incidencia de SG en Latinoamérica.1
Los cinco pacientes que informamos presentaban masa de tejido blando; los casos 1, 3, 4 y 5 también tenían alteraciones orbitarias y de los huesos adyacentes, que se acompañaban de proptosis. El caso 4 presentaba, además, afectación del VII par, causada por una masa en la región inframastoidea. En el caso 2 se pudo evidenciar compromiso de la cara que involucraba el malar y el maxilar, y posiblemente raíces dentarias; por último, el caso 5 tenía afectación del SNC e infiltración de la médula ósea. Los anteriores hallazgos son similares a los descritos en diversas series según las cuales el SG puede afectar huesos, tejidos blandos y la región ocular y producir proptosis, diplopía e inflamación orbitaria.6,11,12
El SG de la órbita es raro y junto con las neoplasias de origen linfoide constituyen el 10% de los tumores orbitarios.13 Las manifestaciones orbitarias ocurren por la infiltración inicial de huesos como el esfenoides y el cigomático; posteriormente el tumor se extiende, más comúnmente a la región lateral de la órbita seguida por la pared superior, el piso orbitario y, por último, la pared media.4
Los hallazgos imaginológicos fueron similares a los previamente descritos en la literatura, a saber: en la TC se encuentran masas isodensas con el músculo, que presentan un realce leve y homogéneo con la administración de contraste intravenoso. En las secuencias T1 de la RM se observan áreas de isointensidad y en T2 hipointensidad relacionada con un alto contenido celular, con realce leve o moderado, algunas veces anular, que puede simular un absceso.14 El compromiso mixto con lesión de tejidos blandos y engrosamiento óseo del maxilar con el que se inició el paciente n.º 2 se considera un hallazgo inusual.
En el diagnóstico diferencial se deben tener en cuenta las siguientes enfermedades:
Rabdomiosarcoma: es el tumor maligno orbitario más frecuente en la primera década de la vida y constituye el 5% de todos los cánceres de la infancia. Su origen es mesenquimal. El subtipo más frecuente es el embrionario. Es agresivo y de crecimiento rápido. Se caracteriza por una masa orbitaria pero es raro que afecte el globo ocular. Más frecuentemente es extraconal, postseptal y superonasal. Puede ocasionar erosión ósea. Usualmente tiene mayor contenido de agua que el SG lo que se manifiesta como lesiones hiperintensas en las secuencias T2 de la RM.15
Histiocitosis de células de Langerhans: procede de la médula ósea y se extiende a la órbita en un 23% de casos. Es frecuente la afectación ósea y característico el aspecto mapiforme de los huesos planos del cráneo; por lo demás su comportamiento imaginológico es similar al del SG.16
Linfoma no Hodgkin de la órbita: representa un 60% a 90% de los linfomas orbitarios. La forma primaria más frecuente es la variedad de células pequeñas tipo B asociada a la mucosa (MALT, por la sigla en inglés de mucosa associated lymphoid tissue) con grado bajo de actividad. Linfoma no Hodgkin de células grandes: es usualmente sistémico y se presenta en adultos. Se manifiesta en general por masas sólidas, homogéneas que captan el contraste y pueden afectar cualquier parte de la órbita. Se moldea al contorno de esta y usualmente es extraconal. Es similar al SG en su contenido altamente celular por lo que se comporta como hipointenso en las secuencias de T2. A diferencia del SG, tiene predilección por la glándula lagrimal y la conjuntiva. Puede infiltrar el músculo simulando la orbitopatía tiroidea.17
Linfoma Burkitt de células T: afecta frecuentemente a los niños.13
Neuroblastoma: en niños se deben tener en cuenta en el diagnóstico diferencial las lesiones secundarias del neuroblastoma; se manifiesta como una masa orbitaria de crecimiento rápido, que afecta el hueso y tiene extensión epidural a través de las suturas.
Lesiones proliferativas linfoides: pueden ser benignas o malignas; la más frecuente es la hiperplasia reactiva o atípica que constituye el 10% a 40% de los casos.18
Seudotumor inflamatorio orbitario: es de carácter idiopático y usualmente ocurre en adultos; a diferencia del SG es doloroso, se acompaña de cambios inflamatorios, y afecta el músculo, la esclerótica y la glándula lacrimal. Por lo demás se comporta en forma similar a las lesiones muy celulares como el SG y el linfoma.19
Hemangiopericitoma: es una lesión sólida, vascular, de bordes irregulares, que destruye el hueso y por ser intensamente vascularizada capta ávidamente el contraste.
Sarcoidosis: a diferencia del SG afecta a la población adulta con predilección por la úvea, la esclerótica, la conjuntiva, la glándula lacrimal, el nervio óptico y las leptomeninges.17
El pronóstico de los enfermos con SG depende de la localización, las estructuras adyacentes afectadas y el comportamiento de la enfermedad de base; el tratamiento es el de esta.4,20,21 La mediana de supervivencia para los pacientes con SG, encontrada en la literatura, es de 4,75 años (rango: 0,1-24 años) pero después del diagnóstico de la enfermedad en la órbita se reduce a 1,5 años (rango: 0,1-7 años) . De 1970 a 1990 la tasa de supervivencia de los pacientes con leucemia mieloide aumentó de 13% a 40% , gracias a los avances en la quimioterapia, la disponibilidad del trasplante de médula ósea y las nuevas modalidades diagnósticas.4
CONCLUSIÓN
El SG es una neoplasia rara que afecta huesos como los de la órbita, el cráneo, el esternón y las costillas, al igual que las regiones proximales de los huesos largos, los tejidos blandos, la piel y los ganglios de pacientes con leucemia mieloide. Las lesiones orbitarias afectan principalmente a niños y jóvenes entre los 5 meses y 18 años. En el contexto de la LMA o las mielodisplasias es la primera posibilidad diagnóstica. Debe hacerse el diagnóstico diferencial, cuando hay lesiones en la cara y el cráneo, con complicaciones de la sinusitis, rabdiomiosarcoma, linfoma de la órbita y otras neoplasias. La imaginología puede ayudar en el diagnóstico diferencial porque demuestra lesiones de los tejidos blandos, usualmente con infiltración, que en algunas oportunidades pueden simular colecciones líquidas como abscesos o invadir el SNC. Es muy característico del SG afectar en un comienzo las paredes lateral y superior de la órbita. Las lesiones orbitarias sugieren que hubo afección primaria de huesos como el esfenoides y el cigomático, con posterior desplazamiento hacia la órbita. Aunque las técnicas imaginológicas ayudan a plantear una hipótesis diagnóstica, su confirmación se debe hacer por histología.
REFERENCIAS BIBLIOGRÁFICAS
1. Fonseca Junior N, Paves L, Nakanami D, Seixa M, Manso P. Sarcoma granulocitico em orbita: relato de caso. Arq Bras Oftalmol (Sao Paulo) 2005; 68: 557-560. [ Links ]
2. Oliva E, Ferry J, Young R, Prat J, Srigley J, Scully R. Granulocytic sarcoma of the female genital tract: A clinicopatologic study of 11 Cases. Am J Surg Pathol 1997; 21: 1156-1165. [ Links ]
3. Deme S, Deodhare S, Tucker W, Bilbao J. Granulocytic sarcoma of the spine in nonleukemic patients: Report of three cases. Neurosurgery 1997; 40: 1283-1287. [ Links ]
4. Bidar M, Wilson M, Laquis S, Wilson T, Fleming J, Wesley R, et al. Clinical and imaging characteristics of orbital leukemic tumors. Ophthal Plast Reconstr Surg 2007; 23: 87-92. [ Links ]
5. Nikolic B, Feigenbaum F, Abbra S, Martuza LR, Schellinger D. CT changes of an intracranial granulocytic sarcoma on short- term follow-up. AJR 2003; 180: 78-80. [ Links ]
6. Manabe Y, Hamakawa Y, Sunami K, Ohta Y, Omori N, Abe K. Granulocytic sarcoma with orbit, cauda equina, muscle and peripheral nerve extension but without bone marrow involvement. Intern Med (Okayama) 2006; 46: 633-635. [ Links ]
7. Bhat VK, Naseeruddin K, Narayanaswamy GN. Sinoorbital chloroma presenting as unilateral proptosis in a boy. Int J Pediatr Otorhinolaryngol 2005; 69:1595-1598. [ Links ]
8. Poblete MT, Zolezzi P, Perez MA, Figueroa C, Zapata C. Sarcoma granulocitico. Rev Chil Pediatr 1984; 55: 188-193. [ Links ]
9. Hernández JA. Sarcoma mieloide: características clínico patológicas y tratamiento. Servicio de Hematología Clínica, Institut Catalá d'Oncologia Hospital Universitari Germans Trias i Pujol Barcelona, España. 2004 Sep. [ Links ]
10. Brunning RD, Matutes E, Harris NL, Flandrin G, Vardiman J, Bennett J, et al. Acute myeloid leukaemia with recurrent genetic abnormalities. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001: 81-87. [ Links ]
11. Blanchard S, Labalette P, Jourdel D, Dedes V, Leleu X, Dillie AF, et al. Sarcome granulocytique orbitaire révélant l'acutisation d'un syndrome myélodysplasique: À propos d'un cas. J Fr Ophtalmol 2004; 27: 184-187. [ Links ]
12. Pomeranz SJ, Hawkins HH, Towbin R, Linsberg WN, Clark RA. Granulocytic sarcoma (chloroma) : CT manifestations. Radiology 1985; 155: 167-170. [ Links ]
13. Valvassori GE, Sabnis SS, Mafee RF, Brown MS, Putterman A. Imaging of orbital lymphoproliferative disorders. Radiol Clin North Am 1999; 37: 135–150. [ Links ]
14. Jung W, Ahn KJ, Park MR, Kim JY, Choi JJ, Kim BS, et al. The radiological spectrum of orbital pathologies that involve the lacrimal gland and the lacrimal fossa. Korean J Radiol (Seoul) 2007;8: 336-342. [ Links ]
15. Conneely MF, Mafee MF. Orbital rhabdomyosarcoma and simulating lesions. Neuroimaging Clin N Am 2005; 15: 121-136. [ Links ]
16. Hidayat AA, Mafee MF, Laver NV, Noujaim S. Langerhans cell histiocytosis and juvenile xanthogranuloma of the orbit. Clinicopathologic, CT, and MR imaging features. Radiol Clin North Am 1998; 36: 1229-1240. [ Links ]
17. Osborn AG, Salzman KL, Wiggins RH, MacDonal A. Orbit, nose and sinuses. En: Harnsberger HR, ed. Diagnostic Imaging Head and Neck, 1st ed. Salt Lake City, Utah: Amirsys; 2004. p. 66-69. [ Links ]
18. Jakobiec FA, McLean I, Font R L. Clinicopathologic charactaristics of orbital lymphoid hyperplasia. Ophthalmology 1979;86: 948-966. [ Links ]
19. Narla LD, Newman B, Spottswood SS, Narla S, Kolli R. Inflammatory pseudotumor. Radiographics 2003; 23: 719-729. [ Links ]
20. Ooi GC, Chim CS, Khong PL, Au WY, Lie AKW, Tsang KWT, et al. Radiologic manifestations of granulocytic sarcoma in adult leukemia. AJR 2001; 176: 1427-1431. [ Links ]
21. Barnet MJ, Zussman WV. Granulocytic sarcoma of the brain: a case report and review of the literature. Radiology 1986; 11: 938-941. [ Links ]
Recibido: septiembre 29 de 2009
Aceptado: noviembre 29 de 2009.

