










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkIatreia
versión impresa ISSN 0121-0793
Iatreia v.23 n.4 Medellín oct./dic. 2010
ARTÍCULO DE REVISIÓN
Sistema NADPH oxidasa: nuevos retos y perspectivas
NADPH oxidase system: new challenges and perspectives
Julián Camilo Arango Rincón1,2,3; Laura Yaneth Gámez Díaz1; Juan Álvaro López Quintero1,2 julian.arango@gmail.com
1 Grupo Inmunodeficiencias primarias, Universidad de Antioquia, Medellín, Colombia.
2 Escuela de Microbiología, Universidad de Antioquia, Medellín, Colombia.
3 Grupo de Microbiología Molecular, Universidad de Antioquia, Medellín, Colombia.
RESUMEN
El sistema NADPH oxidasa es un complejo multiproteico encargado de producir especies reactivas del oxígeno (ROS, por reactive oxygen species) en diferentes células y tejidos. Es de gran importancia en las células fagocíticas (principalmente neutrófilos y macrófagos) porque participa en la destrucción de microorganismos patógenos, mediante la fagocitosis y la formación de las trampas extracelulares de neutrófilos (NET, por neutrophils extracelular traps), así como en la activación de procesos inflamatorios. Las alteraciones en la producción de ROS por parte de las células fagocíticas a causa de defectos genéticos en los componentes del sistema generan la inmunodeficiencia primaria denominada enfermedad granulomatosa crónica (EGC). Este es un artículo de revisión sobre los componentes del sistema NADPH oxidasa, su distribución celular, mecanismo de activación y acción, así como de las funciones que desempeña en otros tejidos. Además, se revisan los defectos moleculares que llevan a la EGC y el tratamiento de esta, incluyendo la terapia con IFNγ, y finalmente las perspectivas para el estudio del sistema.
Palabras clave
Enfermedad granulomatosa crónica, Especies reactivas del oxígeno, NADPH oxidasa, Proteínas NOX
SUMMARY
The NADPH oxidase system is a multiprotein complex that acts as the main source of reactive oxygen species (ROS) in different cells and tissues. In phagocytic cells (mainly macrophages and neutrophils) it is essential for eliminating pathogenic microorganisms, by phagocytosis and the formation of neutrophil extracellular traps (NETs). It also contributes to inflammatory processes. Genetic defects in the components of the system cause alterations in the production of ROS by phagocytic cells, leading to the primary immunodeficiency known as chronic granulomatous disease (CGD). This is a review article on the components of the NADPH oxidase system, its cellular distribution, activation, mechanisms of action, and roles in other tissues. The different molecular defects that lead to EGC are also reviewed, as well as its treatment, including therapy with IFNγ, and the prospects for the study of the system.
Key words Chronic granulomatous disease, NOX proteins, NADPH oxidase system, Reactive oxygen species
INTRODUCCIÓN
Uno de los principales mecanismos inmunes innatos con que cuenta nuestro organismo es el complejo enzimático NADPH oxidasa, cuya función es generar especies reactivas del oxígeno (ROS, por la sigla de reactive oxygen species) encargadas de destruir los gérmenes fagocitados, durante el proceso de estallido respiratorio de las células fagocíticas.1 La importancia de este complejo se evidencia en la enfermedad granulomatosa crónica (EGC), una inmunodeficiencia primaria en la cual la escasez de ROS por parte del sistema NADPH oxidasa ocasiona mayor susceptibilidad a microorganismos oportunistas y al desarrollo de granulomas en diferentes órganos.2 Durante las dos últimas décadas se ha intensificado el estudio del sistema NADPH oxidasa buscando comprender su fisiología, la interrelación con otros mecanismos de inmunidad y su asociación con la EGC. Se han reportado hallazgos interesantes tales como el estado de preactivación (priming) en el sistema NADPH oxidasa, la manifestación de la EGC por defectos en p40phox, el papel de las ROS en la formación de trampas extracelulares de los neutrófilos (NET por la sigla en inglés de neutrophils extracelular traps), un mecanismo microbicida descrito recientemente, y los mecanismos moleculares asociados a la terapia con IFNγ en la EGC. Esta revisión muestra los principales avances en estos temas con el propósito de actualizar los conocimientos acerca del sistema NADPH para comprender mejor el papel de la inmunidad innata en la defensa del organismo.
SISTEMA NADPH OXIDASA
Componentes
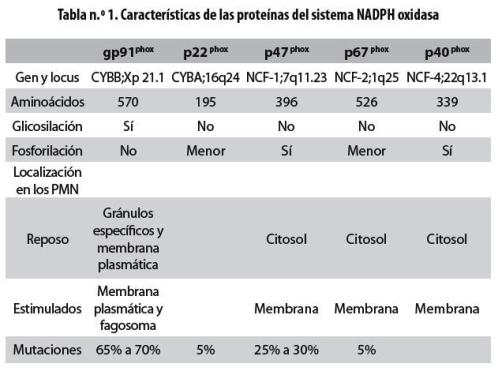
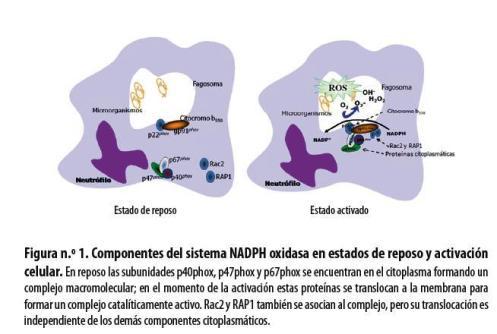
Entre los mecanismos microbicidas de los neutrófilos se encuentra la producción de ROS por parte del sistema NADPH oxidasa, que es un complejo enzimático multiproteico de cinco subunidades distribuidas en el citoplasma, las membranas citoplasmáticas y las vesículas fagocíticas. Los componentes citoplasmáticos son las proteínas p40phox, p47phox y p67phox (la denominación phox es el acrónimo de las palabras inglesas phagocyte oxidase, y el número representa el peso aproximado de cada proteína en KD) que se encuentran formando un complejo macromolecular, mientras que en la membrana se encuentran las proteínas p22phox y gp91phox que conforman un heterodímero llamado flavocitocromo b558 (tabla n.° 1). La activación del complejo enzimático consiste en la translocación de los componentes citoplasmáticos a la membrana de las vacuolas fagocíticas o a la membrana plasmática, mediada por estímulos activadores de la proteína quinasa C (PKC, por la sigla en inglés de protein kinase C) y la participación de proteínas accesorias como Rac2 (GTPasa) y RAP1 (Rasrelated protein 1A) 1 (figura n.° 1).
Componentes de membrana
gp91phox
Es el principal componente del flavocitocromo b 558 y además forma el esqueleto o matriz sobre el cual se ensambla el complejo NADPH oxidasa.1-3 Esta proteína la codifica el gen CYBB cuya extensión es de 30kb, se encuentra en el brazo corto del cromosoma X, región Xp21.1 y posee 13 exones. Está compuesta de 570 aminoácidos con algunas glicosilaciones distribuidas en forma heterogénea en un tercio de su extensión; posee seis dominios de hélices alfa transmembrana que le confieren características hidrofóbicas y tiene como grupos prostéticos dos hemos y un FAD que son importantes para la transferencia de electrones y la unión al NADPH respectivamente.1,2 Como eje central del sistema NADPH oxidasa, gp91phox está involucrada en diferentes procesos celulares entre los que se destacan la destrucción intracelular de microorganismos fagocitados, la liberación de NET, la inducción de muerte celular por apoptosis, la transcripción de diversos genes, el movimiento intermembranal de iones y la secreción de citoquinas y sustancias proinflamatorias.3
Recientemente se ha descrito una nueva clasificación para gp91phox debido al descubrimiento de seis proteínas homólogas que se han denominado NOX1, NOX3, NOX4, NOX5, DUOX1 y DUOX2. La denominación NOX corresponde a NADPH oxidasa y DUOX a dual oxidasa. En general, todas estas moléculas hacen parte de la familia NOX/DUOX del sistema NADPH oxidasa (incluyendo gp91phox que corresponde a NOX2) y se caracterizan por conservar una estructura y función similares a gp91phox. La expresión de estos homólogos ha sido reportada en diferentes tejidos humanos, en otros mamíferos y en plantas.4-7 NOX1se expresa en células del colon, útero, próstata y músculo liso, en los cuales se ha informado una producción escasa de ROS; sin embargo, moléculas proinflamatorias pueden modular positivamente su actividad. 8 NOX3 se expresa fundamentalmente en el riñón fetal y el oído interno.4,5 NOX4, también conocida como Renox, se expresa principalmente en células epiteliales del riñón, así como en osteoclastos, ovarios y ojo;4 finalmente se encuentra NOX5 que, además de tener homología estructural con gp91phox, posee en su extremo amino-terminal un dominio de calmodulina con cuatro sitios de unión para el calcio que le sirve como elemento regulador.9 En relación con el grupo DUOX solo se han encontrado 2 subtipos (DUOX 1 y DUOX2) caracterizados por su doble función oxidasa y peroxidasa.4
p22phox
Es el otro componente del flavocitocromo b558 que también se encuentra anclado a la membrana; la codifica el gen CYBA de 8,5 Kb, que posee seis exones, ubicado en el brazo largo del cromosoma 16, región16q24. La región promotora de p22phox contiene diferentes sitios reguladores como cajas TATA, CCAC y sitios de unión a Sp1, factor nuclear kB y sitios de regulación de interferón gamma.2 Cuenta con 195 aminoácidos, un peso aproximado de 22 KD, tres hélices alfa y un extremo carboxi-terminal orientado hacia la parte citosólica, donde se encuentra una región rica en prolinas que favorece la interacción con p47phox para la estabilidad y ensamblaje del complejo.10,11
Componentes citoplasmáticos
p47phox
Esta proteína la codifica el gen NCF-1 ubicado en la región 7q11.23 del cromosoma 7, con una longitud de 15 Kb y 11 exones; tiene 396 aminoácidos y un peso aproximado de 47 KD. En p47phox se ha descrito un pseudogén con una homología del 97,5%, que corresponde a una copia no funcional caracterizada por presentar una deleción de dos nucleótidos (GT) al comienzo del exón 2.1 Para cumplir su función e interactuar con las otras proteínas del sistema, p47phox cuenta con dos dominios SH3 en el centro de la molécula, un motivo PP en el extremo C-terminal y un domino PX/ PB2 en el N-terminal. Durante la activación del sistema, p47phox es fosforilada aproximadamente en nueve sitios,1 lo cual genera cambios de conformación que le permiten interactuar con p67phox para dar inicio a la translocación de los factores citosólicos a la membrana citoplasmática y de las vacuolas fagocíticas. p47phox cumple esta función porque puede interactuar simultáneamente con proteínas del citoesqueleto (Bactina, miosina) y, además, cumplir el rol de plataforma acopladora de las proteínas citosólicas con las de membrana; por esta razón a p47phox se le ha dado el nombre de NOXO (NADPH oxidase organizer).2,12
p67phox
Esta proteína la codifica el gen NCF-2 ubicado en la región 1q25 del cromosoma 1. Tiene 526 aminoácidos y un peso de 67 KD. Entre sus regiones más importantes se encuentran dos dominios de homología 3 de las Src (SH3), un dominio PB1(Phox and Bem1), un motivo rico en prolinas (PP), cuatro motivos de repeticiones tetrapéptidas (TPR) y tres secuencias de repetición en tándem que son esenciales para interactuar con las otras proteínas del sistema y Rac2 (GTPasa necesaria para la activación). p67phox cumple la función de regular la transferencia de electrones del NADPH al grupo flavina de gp91phox por lo cual ha adquirido el nombre alterno de NOXA (NADPH oxidase activator).4,6
p40phox
El último de los componentes citoplasmáticos del sistema NADPH oxidasa es p40phox codificada por el gen NCF-4 de 18 Kb, que tiene 10 exones y está ubicado en la región 22q 13.1 del cromosoma 22. Pesa 40 KD y tiene 339 aminoácidos; presenta un dominio SH3, un dominio PC en el extremo C-terminal y uno PX/ PB2 en el N-terminal, similar al de p47phox, con el cual puede unir fosfoinositol 3 fosfato (PtdIns[3]P) y otros mediadores importantes en la activación de sistema. En modelos in vitro se ha visto que p40phox no es necesaria para la activación del sistema; sin embargo, sí lo es in vivo para la activación y regulación.13-15
Preactivación y activación del sistema
En estado inactivo las proteínas citosólicas permanecen unidas por las interacciones que se establecen entre los dominios SH3 unidos a motivos ricos en prolina, y los dominios PC unidos a dominios PB1. La activación se caracteriza por un aumento en el consumo de oxígeno molecular y tiene como objetivo la liberación del radical anión superóxido (O2-) dentro de la vacuola fagocítica o al medio extracelular, utilizando como sustrato el fosfato de nicotinamida-adenina dinucleótido (NADPH, por su sigla en inglés). Este fenómeno llamado explosión respiratoria depende del acoplamiento de las proteínas del sistema por cambios de conformación que desenmascaran sitios de unión y permiten que interactúen las unidades citosólicas y el flavocitocromo b558. Cuando se activa el sistema, se producen fosforilaciones en las tres proteínas citosólicas; están mejor documentadas las que se presentan en el extremo C-terminal de p47phox.2 La activación del sistema se desencadena por señales de patógenos opsonizados, citoquinas (IL8, TNFα, endotoxinas [LPS, por lipopolisacárido]), ácido araquidónico, zimozán y factores quimoatrayentes. Estas sustancias tienen como función activar mediadores secundarios intracitoplasmáticos como el PI3k (fosfatidil inositol 3 quinasa) y la PKC (proteína quinasa C), que fosforilan a p47phox, lo cual induce la translocación de las subunidades citosólicas (p40phox, p47phox y p67phox) a la membrana.2,16 Simultáneamente se presenta la translocación de las proteínas Rac2 y RAP1 indispensables para la activación.3,17
Por medio de la superóxido-dismutasa el O2- se transforma en peróxido de hidrógeno (H2O2), que sirve de sustrato a la peroxidasa para oxidar elementos halógenos (Cl-, Br- y I-) y producir ácidos hipohalogenosos. El H2O2 puede además participar en la reacción de Haber-Weiss metal-dependiente para producir el radical hidroxilo (OH-).3,17
En conclusión, el concepto de activación del sistema NADPH oxidasa se refiere a la fosforilación, translocación e interacción completa de sus subunidades luego de un solo estímulo agonista, que se evidencia por la producción de ROS. En 2008 El-Benna y colaboradores2 introdujeron el término priming o estado de preactivación de las células fagocíticas, en el cual el elemento desencadenante no activa completamente el sistema NADPH oxidasa, pero sí hace a la célula más susceptible a una activación posterior por un segundo estímulo, que induce una producción mucho más rápida y fuerte de ROS, debido a que en el momento de la preactivación se fosforila parcialmente p47phox y se acumulan los componentes citoplasmáticos cerca de las membranas, lo cual le permite al segundo estímulo inducir una mejor respuesta.
ENFERMEDAD GRANULOMATOSA CRÓNICA (EGC)
Es una inmunodeficiencia primaria causada por alteraciones génicas en cualquiera de las cinco subunidades proteicas del sistema NADPH oxidasa; es característico de los individuos que la padecen el sufrir infecciones crónicas y granulomas inflamatorios ocasionados por microorganismos oportunistas.18 Se calcula que su frecuencia es de 1/250.000 individuos. Los sitios más frecuentes de infección en estos pacientes son: pulmón, nódulos linfáticos, hígado y tracto gastrointestinal, lo cual genera cuadros clínicos de abscesos en la piel, osteomielitis, otitis media, colitis, enteritis y obstrucción de las vías urinaria y gastrointestinal, causada precisamente por granulomas; en algunos casos se pueden hallar hepatomegalia, esplenomegalia, linfadenitis, anemia, lupus,19 diarrea y sepsis.20,21 Los principales agentes infecciosos asociados con la EGC son microorganismos catalasa positiva como Staphylococcus aureus y Aspergillus fumigatus. Sin embargo, también son de importancia epidemiológica en esta enfermedad otros gérmenes como Salmonella spp, Klebsiella spp, Aerobacter spp, Serratia spp, Pseudomonas spp (Burkholderia cepacia), Aspergillus nidulans, Candida albicans, Scedosporium apiospernum y Chyrosporium zonatum.22
La causa de la EGC es la presencia de mutaciones en cualquiera de los genes que codifican para las proteínas del complejo enzimático NADPH oxidasa de las células fagocíticas. La EGC sigue dos patrones de herencia, uno ligado al cromosoma X (en el que la proteína afectada es gp91phox) y otro autosómico recesivo en caso de alteraciones en los componentes p47phox, p22phox, p67phox y p40phox, por lo que se ha establecido una clasificación basada en los defectos moleculares específicos encontrados. El tipo de herencia se denomina con las letras A para los casos autosómicos y X para los de herencia ligada al sexo. Así mismo, el componente defectuoso del sistema NADPH oxidasa se representa mediante los números 91, 22, 47, 67, o 40 correspondientes a los pesos moleculares de las proteínas afectadas; y los superíndices ''0'', ''+'' o ''–'' que representan los niveles de expresión de las proteínas en cuestión, es decir, proteína ausente, presente o de expresión reducida, respectivamente.23-26
En los pacientes con EGC autosómica (EGC-A) solo el 5% de las mutaciones se presentan en el gen CYBA que codifica para la proteína p22phox; esto la convierte en uno de los casos menos frecuentes de la EGC y también de los más leves debido a que los sitios afectados por lo general no dañan la función catalítica del flavocitocromo b 558.11,27,28 De los defectos autosómicos recesivos, el más común es la deficiencia de p47phox (mutación en el gen NCF-1); se presenta aproximadamente en 30% de los casos.22 A diferencia de la variabilidad de mutaciones relacionadas con los otros componentes del sistema, la mayor parte de las mutaciones en este gen se le atribuyen a una misma causa: una deleción de dos nucleótidos GT al comienzo delexón 2 del gen NCF-1 (del 95% al 97% de los casos presentan esta mutación) lo que trae como consecuencia un ARNm que produce un codón de parada prematuro en el aminoácido 51.29,30
Las mutaciones en la subunidad p67phox son infrecuentes en la EGC-A: solo se presentan en el 5% de los casos;22 los defectos asociados al gen NCF-2 se caracterizan por su gran heterogeneidad lo que provoca inestabilidad en el ARNm y en la proteína. La subunidad p67phox es bastante susceptible a sufrir deleciones y mutaciones sin sentido (missense) lo cual provoca la sustitución de aminoácidos en su extremo N-terminal; esto dificulta considerablemente el funcionamiento de la proteína, pues en esta zona se encuentran sus componentes funcionales y se dan las principales interacciones con la p47phox y las proteínas Rac2 y RAP1.31,32
Hasta hace pocos años no se tenía claro el papel de p40phox en el funcionamiento del sistema NADPH oxidasa y no se la había llegado a asociar con la EGC-A. El primer informe de EGC-A por mutaciones en NCF-4 (que codifica para p40phox) lo publicaron en 2009 Matute y colaboradores13; se trataba de un niño con colitis granulomatosa, cuyos neutrófilos mostraban un defecto sustancial en la producción de anión superóxido en los fagosomas, mientras que la liberación extracelular de ROS no se encontró afectada. El análisis genético mostró dos mutaciones, a saber: una que genera un cambio de sentido que da origen a un codón de parada prematura, y la otra, una mutación sin sentido que induce una sustitución R105Q en el dominio PX, que le impide la unión a dominios PtdIns(3)P, por lo cual no se da la producción de ROS en el fagosoma.13,14 Estos hallazgos evidencian la importancia de p40phox en la activación del sistema NADPH oxidasa y generan la necesidad de evaluar no solo la producción extracelular de ROS, como lo hacen la mayoría de las técnicas, sino también la intracelular en el fagosoma.
El patrón ligado al X (EGC-X) es el más frecuente pues representa el 60% de los casos de EGC.20 El fenotipo común es el X910 y se refiere a la ausencia del citocromo b558 y a la actividad nula de NADPH oxidasa. El fenotipo X91- es menos frecuente y se refiere a una forma variante de la EGC caracterizada por actividad baja de NADPH oxidasa, proporcional al nivel de citocromo b558 expresado. En el fenotipo X91+ el citocromo b558 se encuentra en el nivel normal, pero su actividad está disminuida o ausente.28 Las mujeres heterocitogóticas para la enfermedad presentan un porcentaje pequeño de producción de ROS de 5 a 10% en la prueba de reducción del azul de tetrazolio (NBT, por la sigla en inglés de nitroblue tetrazolium) y en su mayoría son asintomáticas, sin embargo un grupo reducido de ellas presenta infecciones recurrentes y enfermedades autoinmunes, principalmente lupus.19,33,34
Entre las mutaciones que se presentan en CYBB se encuentran las asociadas a la región promotora (2%), las deleciones o inserciones grandes (14%) y las deleciones o inserciones pequeñas (26%); el 58% restante lo constituyen las mutaciones puntuales, las mutaciones sin sentido o con cambio de sentido que pueden llevar a alteraciones en el procesamiento del ARNm y las mutaciones que alteran los sitios de empalme (splicing).25,28
Aunque las alteraciones de la proteína Rac2 no llevan a EGC, se han informado pacientes con mutaciones en ella que presentan infecciones recurrentes.35
De otro lado, cabe anotar que un defecto en la explosión respiratoria causa un bloqueo en la producción de las trampas extracelulares de los neutrófilos (NET), estructuras compuestas por cromatina y proteínas granulares, cuya función es atrapar extracelularmente bacterias grampositivas, gramnegativas y hongos, con el fin de promover su destrucción.36,37 Diferentes autores proponen que las ROS intervienen como segundos mensajeros para la síntesis de las NET, por lo que se ha planteado que las consecuencias adversas de la EGC no se deben solo a la ausencia de capacidad microbicida de las ROS, sino también a defectos en la producción de NET.36-38 Bianchi y colaboradores demostraron la importancia de las NET en la EGC, pues al hacer terapia génica no solo consiguieron recuperar la capacidad oxidasa del sistema sino que, además, las células recuperaron la producción de NET y la capacidad microbicida.38 A este respecto se necesitan estudios que permitan comprender el verdadero papel de las NET en la EGC y la forma en que se pueda restablecer o modular.
Es importante mencionar que la mayoría de los pacientes con EGC-X tienen mutaciones exclusivas de sus familias lo cual explica la heterogeneidad clínica y genética de la enfermedad. En este sentido, el estudio además de ilustrar la importancia clínica de las ROS, ha permitido identificar los diversos componentes del sistema NADPH oxidasa, así como sus mecanismos de activación y las formas posibles de tratamiento.22
DIAGNÓSTICO DE LA ENFERMEDAD GRANULOMATOSA CRÓNICA
La sospecha de un caso de EGC se basa principalmente en las señales de alerta que, de acuerdo con el PAGID (por la sigla en inglés de Panamerican Group for Immunodeficiencies), consisten en dos o más episodios de adenitis que requieran drenaje quirúrgico, neumonía por hongos oportunistas, abscesos hepáticos por S. aureus o Aspergillus, efectos adversos de la vacuna BCG, infección grave por S. aureus, Serratia marcescens, B. cepacia, Pseudomonas spp, Aspergillus spp, Candida spp y Nocardia spp, y una historia familiar de infecciones repetidas. El diagnóstico de laboratorio se fundamenta en pruebas bioquímicas que evalúan la capacidad de los neutrófilos de producir anión superóxido y otras ROS; entre ellas se cuentan los ensayos cualitativos como el NBT y las pruebas cuantitativas mediante citometría de flujo utilizando dihidrorrodamina (DHR) o reducción del citocromo C.39-41 Cuando las pruebas se hacen simultáneamente al paciente y su madre (en el caso de pacientes hombres) es posible deducir el patrón hereditario de la enfermedad.
Después de confirmar el diagnóstico mediante las pruebas mencionadas, se procede a los estudios genéticos de los pacientes y sus familiares; la mayor parte de las técnicas para la búsqueda de mutaciones se basan en la reacción en cadena de la polimerasa (PCR) a partir de ADN genómico (ADNg) o de ADN copia (ADNc); se continúa con ensayos de polimorfismo conformacional de cadena simple (SSCP, por la sigla en inglés de single strand conformation polymorphism) y secuenciamiento.39,42
TRATAMIENTO DE LA ENFERMEDAD GRANULOMATOSA CRÓNICA
El tratamiento actual de los pacientes con EGC se basa en el control de los procesos infecciosos y la profilaxis antimicrobiana.23 La administración de interferón gamma humano recombinante (IFNγ) está indicada como tratamiento profiláctico e inmunomodulador y con él se ha demostrado una disminución de la recurrencia y gravedad de las infecciones. Se administra por vía subcutánea tres veces por semana, en dosis de 50 μg/m2 de superficie corporal; es capaz de reducir en un 70% el riesgo relativo de infecciones graves.23,43
El IFNγ induce la expresión del gen CYBB y la diferenciación monocítica de progenitores.44,45 Varios mecanismos de regulación se asocian a esta citoquina; entre ellos se encuentra que induce la unión de BID/YY1 (binding increased differentiation, factor de transcripción Yin Yang, Yin Yang transcription factor) a cuatro sitios en la región –90 a –355 pb del promotor después de la proteína de desplazamiento CDP (por la sigla de CCAAT displacement protein) del gen CYBB;46 otro se basa en la unión de un complejo multiproteico no definido (PU.1/HAF-1) localizado a –53 del promotor 47,48; además existe un mecanismo, descrito por Kumatori y colaboradores, que propone la formación de un gran complejo compuesto por los transductores de señal y activadores de transcripción 1α (STAT-1α, por la sigla de signal transducers and activators of transcription 1α), el factor regulador del interferón-1 (IRF-1, por la sigla de interferon regulatory factor-1), el factor de transcripción que reconoce secuencias ricas en purinas (PU.1), la proteína de unión a la secuencia consenso del IFN (ICSBP, por la sigla de IFN consensus-binding protein) y la proteína fijadora de CREB (CBP, por la sigla de CREB-binding protein) en el promotor proximal de CYBB después del estímulo con IFN-γ.49 Que el IFN-γ sea capaz de inducir la expresión del gen CYBB sirvió, en parte, de base para usarlo clínicamente en la prevención de infecciones en pacientes con EGC.50
El mecanismo de acción del IFN-γ no está totalmente esclarecido pero se sabe que se basa en la estimulación del sistema NADPH oxidasa, principalmente porque induce una mayor expresión de gp91phox. Esto explica su mayor eficacia en las formas ligadas al cromosoma X y específicamente en las variantes relacionadas con el empalme (splicing).43 En tal sentido, se ha reportado que el uso del IFNγ mejora la fidelidad del splicing,51 generando transcriptos alternos mediante el fenómeno de splicing alternativo.51,52 Sin embargo, a pesar de este conocimiento, aún no están totalmente claros los mecanismos moleculares desencadenados por el IFN-γ en la regulación del empalme.53,54
Otra de las propuestas para el tratamiento de esta inmunodeficiencia es el trasplante de médula ósea, pero hasta el momento no existe acuerdo sobre este enfoque curativo, porque tiene limitaciones importantes, entre ellas: la dificultad de encontrar un donante histocompatible, la alta probabilidad de desarrollar enfermedad injerto contra hospedero (EIH) y la susceptibilidad a microorganismos oportunistas dada la inmunosupresión generada por las condiciones mieloablativas previas al trasplante.23,55,56
Finalmente, otra opción curativa para la EGC es la terapia génica, que constituye un buen modelo para explorar diversas estrategias en la incorporación de transgenes a células mieloides.57 Uno de los principales ensayos clínicos de terapia génica se hizo en cinco pacientes con deficiencia de p47phox de quienes se obtuvieron células CD34+ movilizadas a la sangre periférica, a las cuales se les introdujo un vector retroviral que contenía el ADNc de p47phox, para luego reperfundirlas a los pacientes. Este trabajo mostró una restauración de 6% a 29% de la actividad oxidasa en células diferenciadas in vitro en granulocitos obtenidos a partir de las células CD34+ transducidas; sin embargo, se presentó un bajo porcentaje de neutrófilos corregidos en sangre periférica, entre 0,004% y 0,05% después de la transfusión de dichas células.58,59 Este estudio y otros similares reflejan una baja eficiencia de la transferencia génica mediada por retrovirus en células progenitoras hematopoyéticas humanas. Es interesante observar que en estudios llevados a cabo con modelos animales bajo las mismas condiciones y con el mismo protocolo, se obtiene un 5% de células de sangre periférica con actividad oxidasa después del trasplante, lo que indica que la falla radica en la falta de optimización de los procedimientos de terapia génica aplicados en células humanas; por lo tanto, se espera que con nuevas técnicas basadas en liposomas, o mediante la nanotecnología, se pueda finalmente corregir, en forma parcial o total, el gen CYBB.57
PERSPECTIVAS EN EL ESTUDIO DEL SISTEMA NADPH OXIDASA
Como se dijo anteriormente, el sistema NADPH oxidasa se expresa y tiene una alta funcionalidad principalmente en células fagocíticas como los neutrófilos; por lo tanto, para su estudio es necesario usar este tipo de células; infortunadamente los aislados hematopoyéticos de neutrófilos humanos y murinos tienen la gran desventaja de ser células maduras difíciles de conservar y manipular genéticamente. Con el uso de líneas hematopoyéticas inmortales ha sido posible estudiarlo, pero en estos modelos han sido muy reducidos el nivel de producción de anión superóxido y la expresión de las proteínas transgénicas.60 Otra posibilidad para el estudio del sistema NADPH oxidasa, que ha generado información importante acerca de las proteínas que lo componen, es la de usar modelos artificiales o libres de células pero, como es de suponer, existen grandes diferencias entre la reconstitución artificial y el sistema in vivo.61
El modelo COS-7 consiste en una línea celular originada de células de riñón de mono verde africano, que supera las limitaciones de los modelos ya mencionadas porque es transgénica para las proteínas del sistema NADPH oxidasa, y muestra una alta expresión de estos componentes y producción significativa de O2 - ante estímulos como el forbol miristato acetato (PMA, por la sigla en inglés de phorbol myristate acetate, y el ácido araquidónico); estas son sustancias que activan mediadores como la proteína quinasa C (PKC, por la sigla en inglés de protein kinase C) y Rac2, los mismos que se activan en las células fagocíticas. En las células COS-7 se puede suprimir cualquiera de los genes participantes en el sistema NADPH oxidasa, para luego transfectar el respectivo gen mutante permitiendo crear in vitro un modelo alterado del sistema NADPH que puede informar acerca de su activación y de la expresión e interacciones de sus proteínas.31,61
CONCLUSIÓN
El estudio del sistema NADPH oxidasa de las células fagocíticas durante los últimos años se ha diversificado con los avances de las técnicas inmunológicas y moleculares, lo cual ha permitido conocer diferentes proteínas homólogas del sistema en otros tejidos y otros seres vivos, la forma en que interactúan los componentes y los procesos involucrados en la preactivación y activación del sistema. Además, cada vez se le atribuyen más funciones a este sistema que van desde la generación de NET hasta la regulación de la expresión génica y la activación de algunas vías de apoptosis. Este conocimiento amplio del sistema NADPH oxidasa a su vez ha permitido entender mejor la patogénesis de la EGC, en la que uno de los hallazgos más interesantes son los defectos en p40phox. Así mismo, la comprensión de las vías moleculares y los mecanismos de acción del IFNγ genera un panorama alentador en el desarrollo de terapias más seguras, eficaces y específicas de cada alteración.
REFERENCIAS BIBLIOGRÁFICAS
1. Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci, 2002; 59 (9): 1428-1459. [ Links ]
2. El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol, 2008; 30 (3): 279-289. [ Links ]
3. Rada B, Hably C, Meczner A, Timar C, Lakatos G, Enyedi P, et al. Role of Nox2 in elimination of microorganisms. Semin Immunopathol, 2008; 30 (3): 237-253. [ Links ]
4. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev, 2007; 87 (1): 245-313. [ Links ]
5. Segal AW. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int J Biochem Cell Biol, 2008; 40 (4): 604-618. [ Links ]
6. Behe P, Segal AW. The function of the NADPH oxidase of phagocytes, and its relationship to other NOXs. Biochem Soc Trans, 2007; 35 (Pt. 5): 1100-1103. [ Links ]
7. Kawahara T, Lambeth JD. Molecular evolution of Phox-related regulatory subunits for NADPH oxidase enzymes. BMC Evol Biol, 2007; 7 (178): 1-29. [ Links ]
8. Katsuyama M, Fan C, Yabe-Nishimura C. NADPH oxidase is involved in prostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells: induction of NOX1 by PGF2alpha. J Biol Chem, 2002; 277 (16): 13438-13442. [ Links ]
9. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol, 2004; 4 (3): 181-189. [ Links ]
10. Miyano K, Sumimoto H. Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie, 2007; 89 (9): 1133-1144. [ Links ]
11. Tahara T, Shibata T, Wang F, Nakamura M, Sakata M, Nakano H, et al. A genetic variant of the p22phox component of NADPH oxidase C242T is associated with reduced risk of functional dyspepsia in Helicobacter pylori-infected Japanese individuals. Eur J Gastroenterol Hepatol, 2009; 21 (12): 1363-1368. [ Links ]
12. El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med, 2009; 41: 217-225. [ Links ]
13. Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil NADPH oxidase activity. Blood, 2009; 114 (15): 3309-3315. [ Links ]
14. Matute JD, Arias AA, Dinauer MC, Patino PJ. p40phox: the last NADPH oxidase subunit. Blood Cells Mol Dis, 2005; 35: 291-302. [ Links ]
15. Ueyama T, Tatsuno T, Kawasaki T, Tsujibe S, Shirai Y, Sumimoto H, et al. A regulated adaptor function of p40phox: distinct p67phox membrane targeting by p40phox and by p47phox. Mol Biol Cell, 2007; 18 (2): 441-454. [ Links ]
16. Singh A, Zarember KA, Kuhns DB, Gallin JI. Impaired priming and activation of the neutrophil NADPH oxidase in patients with IRAK4 or NEMO deficiency. J Immunol, 2009; 182 (10): 6410-6417. [ Links ]
17. Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol, 2008; 15: 164-187. [ Links ]
18. Johnston RB, Jr. Clinical aspects of chronic granulomatous disease. Curr Opin Hematol, 2001; 8: 17-22. [ Links ]
19. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol, 2007; 148: 79-84. [ Links ]
20. Elloumi HZ, Holland SM. Diagnostic assays for chronic granulomatous disease and other neutrophil disorders. Methods Mol Biol, 2007; 412: 505-523. [ Links ]
21. Rosenzweig SD. Inflammatory manifestations in chronic granulomatous disease (CGD). J Clin Immunol, 2008; 28 (Suppl. 1): S67-72. [ Links ]
22. Stasia MJ, Li XJ. Genetics and immunopathology of chronic granulomatous disease. Semin Immunopathol, 2008; 30 (3): 209-235. [ Links ]
23. Seger RA. Modern management of chronic granulomatous disease. Br J Haematol, 2008; 140 (3): 255-266. [ Links ]
24. Winkelstein JA, Marino MC, Johnston RB, Jr., Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore), 2000; 79 (3): 155-169. [ Links ]
25. Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol, 2003; 15 (5): 578-584. [ Links ]
26. Stasia MJ, Bordigoni P, Floret D, Brion JP, Bost-Bru C, Michel G, et al. Characterization of six novel mutations in the CYBB gene leading to different sub-types of X-linked chronic granulomatous disease. Hum Genet, 2005; 116 (1-2): 72-82. [ Links ]
27. Feairheller DL, Brown MD, Park JY, Brinkley TE, Basu S, Hagberg JM, et al. Exercise training, NADPH oxidase p22phox gene polymorphisms, and hypertension. Med Sci Sports Exerc, 2009; 41 (7): 1421-1428. [ Links ]
28. Rae J, Noack D, Heyworth PG, Ellis BA, Curnutte JT, Cross AR. Molecular analysis of 9 new families with chronic granulomatous disease caused by mutations in CYBA, the gene encoding p22(phox). Blood, 2000; 96 (3): 1106-1112. [ Links ]
29. Noack D, Rae J, Cross AR, Ellis BA, Newburger PE, Curnutte JT, et al. Autosomal recessive chronic granulomatous disease caused by defects in NCF-1, the gene encoding the phagocyte p47-phox: mutations not arising in the NCF-1 pseudogenes. Blood, 2001; 97 (1): 305-311. [ Links ]
30. Heyworth PG, Noack D, Cross AR. Identification of a novel NCF-1 (p47-phox) pseudogene not containing the signature GT deletion: significance for A47 degrees chronic granulomatous disease carrier detection. Blood, 2002; 100 (5): 1845-1851. [ Links ]
31. Arias AA, Dinauer MC, Ding J, Matute JD, Patino PJ. Expression and activity of polymorphisms in the 67- kDa protein of the NADPH oxidase system. Biomedica, 2004; 24 (3): 262-272. [ Links ]
32. Grizot S, Fieschi F, Dagher MC, Pebay-Peyroula E. The active N-terminal region of p67phox. Structure at 1.8 A resolution and biochemical characterizations of the A128V mutant implicated in chronic granulomatous disease. J Biol Chem, 2001; 276 (24): 21627-21631. [ Links ]
33. Aguilera P, Mascaro JM, Jr., Martinez A, Esteve J, Puig S, Campo E, et al. Cutaneous gamma/delta T-cell lymphoma: a histopathologic mimicker of lupus erythematosus profundus (lupus panniculitis). J Am Acad Dermatol, 2007; 56 (4): 643-647. [ Links ]
34. Cordoba-Guijarro S, Feal C, Dauden E, Fraga J, Garcia- Diez A. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol, 2000; 14 (5): 409-411. [ Links ]
35. George A, Pushkaran S, Li L, An X, Zheng Y, Mohandas N, et al. Altered phosphorylation of cytoskeleton proteins in sickle red blood cells: The role of protein kinase C, Rac GTPases, and reactive oxygen species. Blood Cells Mol Dis, 2010; 45 (1): 41-45. [ Links ]
36. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol, 2007; 176 (2): 231-241. [ Links ]
37. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science, 2004; 303 (5663): 1532-1535. [ Links ]
38. Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood, 2009; 114 (13): 2619-2622. [ Links ]
39. Jirapongsananuruk O, Niemela JE, Malech HL, Fleisher TA. CYBB mutation analysis in X-linked chronic granulomatous disease. Clin Immunol, 2002; 104 (1): 73-76. [ Links ]
40. Wu YC, Huang YF, Lin CH, Shieh CC. Detection of defective granulocyte function with flow cytometry in newborn infants. J Microbiol Immunol Infect, 2005; 38 (1): 17-24. [ Links ]
41. Lehmann AK, Sornes S, Halstensen A. Phagocytosis: measurement by flow cytometry. J Immunol Methods, 2000; 243 (1-2): 229-242. [ Links ]
42. Agudelo-Florez P, Prando-Andrade CC, Lopez JA, Costa-Carvalho BT, Quezada A, Espinosa FJ, et al. Chronic granulomatous disease in Latin American patients: clinical spectrum and molecular genetics. Pediatr Blood Cancer, 2006; 46 (2): 243-252. [ Links ]
43. Errante PR, Frazao JB, Condino-Neto A. The use of interferon-gamma therapy in chronic granulomatous disease. Recent Pat Antiinfect Drug Discov, 2008; 3 (3): 225-230. [ Links ]
44. Cassatella MA, Hartman L, Perussia B, Trinchieri G. Tumor necrosis factor and immune interferon synergistically induce cytochrome b-245 heavy-chain gene expression and nicotinamide-adenine dinucleotide phosphate hydrogenase oxidase in human leukemic myeloid cells. J Clin Invest, 1989; 83 (5): 1570-1579. [ Links ]
45. Newburger PE, Ezekowitz RA, Whitney C, Wright J, Orkin SH. Induction of phagocyte cytochrome b heavy chain gene expression by interferon gamma. Proc Natl Acad Sci USA, 1988; 85 (14): 5215-5219. [ Links ]
46. Jacobsen BM, Skalnik DG. YY1 binds five cis-elements and trans-activates the myeloid cell-restricted gp91(phox) promoter. J Biol Chem, 1999; 274 (42): 29984-29993. [ Links ]
47. Eklund EA, Jalava A, Kakar R. PU.1, interferon regulatory factor 1, and interferon consensus sequencebinding protein cooperate to increase gp91(phox) expression. J Biol Chem, 1998; 273 (22): 13957-13965. [ Links ]
48. Eklund EA, Kakar R. Recruitment of CREB-binding protein by PU.1, IFN-regulatory factor-1, and the IFN consensus sequence-binding protein is necessary for IFN-gamma-induced p67phox and gp91phox expression. J Immunol, 1999; 163 (11): 6095-6105. [ Links ]
49. Kumatori A, Yang D, Suzuki S, Nakamura M. Cooperation of STAT-1 and IRF-1 in interferon-gammainduced transcription of the gp91(phox) gene. J Biol Chem, 2002; 277 (11): 9103-9111. [ Links ]
50. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The International Chronic Granulomatous Disease Cooperative Study Group. N Engl J Med, 1991; 324 (8): 509-516 [ Links ]
51. Condino-Neto A, Newburger PE. Interferon-gamma improves splicing efficiency of CYBB gene transcripts in an interferon-responsive variant of chronic granulomatous disease due to a splice site consensus region mutation. Blood, 2000; 95 (11): 3548-3554. [ Links ]
52. Weening RS, de Klein A, de Boer M, Roos D. Effect of interferon-gamma, in vitro and in vivo, on mRNA levels of phagocyte oxidase components. J Leukoc Biol, 1996; 60 (6): 716-720. [ Links ]
53. Errante PR, Prando C, Bustamante J, Aragao Filho WC, Pereira PV, Rehder J, et al. Comment on ''Impaired priming and activation of the neutrophil NADPH oxidase in patients with IRAK4 or NEMO deficiency''. J Immunol, 2009; 183 (6): 3559. [ Links ]
54. Luengo-Blanco M, Prando C, Bustamante J, Aragao- Filho WC, Pereira PV, Rehder J, et al. Essential role of nuclear factor-kappaB for NADPH oxidase activity in normal and anhidrotic ectodermal dysplasia leukocytes. Blood, 2008; 112 (4): 1453-1460. [ Links ]
55. Soncini E, Slatter MA, Jones LB, Hughes S, Hodges S, Flood TJ, et al. Unrelated donor and HLA-identical sibling haematopoietic stem cell transplantation cure chronic granulomatous disease with good long-term outcome and growth. Br J Haematol, 2009; 145 (1): 73-83. [ Links ]
56. Sokolic R, Kesserwan C, Candotti F. Recent advances in gene therapy for severe congenital immunodeficiency diseases. Curr Opin Hematol, 2008; 15 (4): 375-380. [ Links ]
57. Velásquez LA, Arango JC, Arias AA, Patiño PJ. Importancia de la terapia génica en la enfermedad granulomatosa crónica. Iatreia, 2005; 18 (3): 308-319. [ Links ]
58. Mardiney M, 3rd, Jackson SH, Spratt SK, Li F, Holland SM, Malech HL. Enhanced host defense after gene transfer in the murine p47phox-deficient model of chronic granulomatous disease. Blood, 1997; 89 (7): 2268-2275. [ Links ]
59. Malech HL, Maples PB, Whiting-Theobald N, Linton GF, Sekhsaria S, Vowells SJ, et al. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci USA, 1997; 94 (22): 12133-12138. [ Links ]
60. Arango JC, Arias AA, Patiño PJ. Importancia de los ratones knock-out en el estudio del sistema NADPH oxidasa. Rev Cubana Hematol Inmunol Med Transfus, 2005; 21 (2): 1-10. [ Links ]
61. Pick E, Bromberg Y, Shpungin S, Gadba R. Activation of the superoxide forming NADPH oxidase in a cell-free system by sodium dodecyl sulfate. Characterization of the membrane-associated component. J Biol Chem, 1987; 262 (34): 16476-16483. [ Links ]
Recibido: noviembre 17 de 2009
Aceptado: marzo 22 de 2010

