










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkIatreia
versão impressa ISSN 0121-0793
Iatreia v.24 n.2 Medellín abr./jun. 2011
INVESTIGACIÓN ORIGINAL
Purification and activation of caprine and canine plasminogens: Comparison with human plasminogen
Purificación y activación de los plasminógenos caprino y canino: comparación con el plasminógeno humano
Omaira Cañas Bermúdez 1; Alfonso Quijano Parra1; Luis Fernando Arbeláez Ramírez2
1 Grupo de Investigación en Química, Universidad de Pamplona, Pamplona, Colombia
2 Grupo de Investigación en Química, Universidad de Pamplona, Pamplona, Colombia, luifer@unipamplona.edu.co.
SUMMARY
Objective: To unify the purification and activation of plasminogens from three different species, namely: human, caprine and canine. Materials and methods: Lysine-Sepharose 4B and sephacel DEAE were used, for affinity and ion-exchange chromatography, respectively. The N-terminal sequence was determined for both the intact and degraded plasminogens. Results: Bands of 92 kDa corresponding to native plasminogens were identified in the three species. Their N-terminal sequences were found to be EPLDDY, DPLDDY and XXLDDY for human, caprine and canine plasminogen, respectively. Furthermore, the degraded in vivo circulating plasminogens from the three species were purified and their N-terminal sequences were KVYLSE, RITLL and RIYLS for the human, caprine and canine, in that order. Conclusion: Activation of the three plasminogens confirmed the formation of the typical electrophoretic bands for human plasmin corresponding to the heavy A and the light B chains which were also identified in the caprine and canine plasmins. This new purification methodology facilitates the comparison and further elucidation of the fibrinolytic systems in mammals.
Key Words
Blood Coagulation Tests, Plasminogen, Tissue Plasminogen Activator
RESUMEN
Objetivo: unificar la purificación y activación de los plasminógenos de tres especies diferentes, a saber: humana, caprina y canina. Materiales y métodos: se usaron Lysina-Sefarosa 4B y Sefacel DEAE para las cromatografías de afinidad y de intercambio iónico, respectivamente. Se determinó la secuencia terminal-N tanto de los plasminógenos intactos como de los degradados. Resultados: en las tres especies se identificaron bandas de 92 kDa correspondientes a los plasminógenos nativos. Se halló que sus secuencias terminales-N eran EPLDDY, DPLDDY y XXLDDY para los plasminógenos humano, caprino y canino, respectivamente. Además, se purificaron los plasminógenos degradados circulantes, cuyas secuencias terminales-N fueron, en el mismo orden, KVYLSE, RITLL Y RIYSL. Conclusión: la activación de los tres plasminógenos confirmó la formación de las bandas electroforéticas típicas de la plasmina humana correspondientes a las cadenas pesada A y liviana B, que también se identificaron en las plasminas caprina y canina. Este nuevo método de purificación facilita la comparación y el esclarecimiento de los sistemas fibrinolíticos de los mamíferos.
Palabras clave
Pruebas de Coagulación Sanguínea, Plasminógeno, Activador de Tejido Plasminógeno
INTRODUCTION
Haemostasis is the arrest of hemorrhage and a response to vascular injury; it involves vasoconstriction, tissue swelling, coagulation and thrombosis (1). Coagulation of blood is mediated by cellular components and soluble plasma proteins in response to vascular injury. In the final step, thrombin cleavages fibrinogen to generate fibrin monomers that polymerize to form a chemically stable clot.
The fibrinolytic (plasminogen/plasmin) system in the vasculature includes an inactive proenzyme, plasminogen (Pg), which can be converted into the active enzyme, plasmin (Pm), which degrades fibrin into soluble degradation products (2). Two immunologically distinct plasminogen activators (PA) have been identified: the tissue-type PA (t-PA) and the urokinase-type PA (u-PA). The t-PA mediated Pg activation is mainly involved in the dissolution of fibrin in the circulation. Inhibition of the fibrinolytic system may occur either at the level of PA by specific Pg activator inhibitors, types 1 and 2 (PAI-1 and PAI-2), or at the level of Pm; this is done mainly by α2-antiplasmin (α2-AP) (3,4). As with most other plasma proteins, Pg is synthesized in the liver and the human plasma concentration is approximately 2 µM/L (5); native Pg is a single-chain glycoprotein with glutamic acid as the N-terminal for human Pg (6), and is therefore referred to as Glu-Pg. It is, however, easily degraded and autocatalytically cut by Pm; this releases a peptide of 8 kDa by cleavage at Lys76-Lys77 that converts Glu-Pg into Lys77-Pg (7). Calculations based on the primary sequence, corrected for carbohydrates (8,9) give molecular weights of 92 kDa and 82 kDa for Glu- and Lys-Pg respectively (5).
Through cleavage of a single peptide bond (Arg560-Val561), Pg is activated to Pm (10). This slicing is equivalent to the activation cleavage of other serine enzymes and the two chains are held together by two disulfide bonds (11). The full length cDNA of human Pg has been cloned and analysis of the DNA sequence has revealed that Pg contains 791 amino-acid residues (12).
The Pg molecule contains lysine binding sites (LBS) to which lysine or its analogues carrying both an amino group and a free carboxyl group bind; among them: arginine and epsilon amino caproic acid (εACA). One strong and five weak LBS were found (13), thus assuming that one of the low-affinity sites represents the active one. The other five sites correspond to the five kringle domains in the Pg A-chain (11). The binding of Pg to fibrin, α2-AP, histidine rich glycoprotein and thrombospondin is mediated through the kringles (14-16).
Plasminogens from several species have been isolated and studied. So far, human Pg has been the most thoroughly analyzed. It has been purified by different methods and multiple molecular forms of human Pg have been identified and structurally analyzed (17).
Properties of Pgs from other mammalian species, notably cows (18), rabbits and sheep (19) have so far been investigated and compared to human Pg. It has been well established that Pgs from distinct species differ in behaviour towards streptokinase. Human, cat and monkey Pgs are readily activated by catalytic amounts of streptokinase, whereas the opposite occurs in bovine, pig, sheep, rat and mouse Pgs (20). The proenzymes from dogs and rabbits require high concentrations of streptokinase for activation. Human Pg and Pm form a stoichiometric 1:1 complex with streptokinase (21).
In this report we present the results of a simplified method for purifying Pgs from three different species, namely: human, caprine and canine. Of them, caprine Pg has now been purified for the first time, and purification of canine Pg has been improved. Pg concentration in these three species was compared, and their N-terminal sequences were determined and compared.
MATERIALS AND METHODS
Chemicals
All buffers, salts and other chemicals were of the highest available purity: εACA, phenylmethanesulfonyl fluoride (PMSF), dimethylsulfoxide (DMSO), methanol and acetic acid were from Fluka. N, N-methylenebis- acrylamide, ammonium persulfate, N,N,N',N'- tetramethylethylenediamine, β-mercaptoethanol and sodium dodecyl sulfate (SDS) were from BioRad. Sodium chloride (NaCl) and sodium acetate were produced by Riedel-de Haën. Di-sodium hydrogen phosphate dihydrate was from Merck, and Lysine- Sepharose® 4B was supplied by Amersham Biosciences. Diethylaminoethyl (DEAE) and aprotinin were supplied by Sigma. Chromogenic substrate for Pm Spectrozyme and Urokinase (Uk) were from American Diagnostica Inc. The Spectrolyze® Plasminogen SK kit was supplied by Trinity Biotech. The molecular weight markers employed (180 kDa [α2-macroglobulin], 92 kDa [Glu-Pg], 66 kDa [α-chain human fibrinogen], 52 kDa [β-chain human fibrinogen], 46 kDa [γ-chain human fibrinogen] and 23 kDa [Trypsin] were supplied by Laboratorios de Investigationes en Biomoléculas from Pamplona University (Pamplona, Colombia).
Plasma samples
Both human and animal bloods were drawn into bags containing 0.13 mol/L trisodium citrate as anticoagulant. Human Pg was obtained from fresh plasma supplied by the Erasmo Meoz Hospital (Cúcuta, Colombia); before use, it was analyzed and certified to be free from antigens of hepatitis, VIH, Chagas and other infectious diseases. Animal blood was obtained from the Experimental farm Villa Marina (Pamplona University, Colombia). To 200 mL of plasma from each species 1mM PMSF (disolved in DMSO) and 730 IU/ml Aprotinin were added as serinoproteinases inhibitor.
Affinity chromatography
All Pgs were purified by affinity chromatography on Lysine-Sepharose® 4B, according to the method of Deutsch and Mertz (22), using 35 mL of Lysine- Sepharose® 4B packed in a column of 12 x 2.0 cm from BioRad, equilibrated with three column volumes of 0.1 M phosphate buffer containing 0.15 M of NaCl pH 7.3 (PBS) at a flow rate of 2 mL/min followed by the application of the plasma sample (200 mL) and washed with the same buffer until obtaining the absorbance A280 at ≤ 0.01. The bound Pgs were eluted with 100 mL of PBS containing 0.05 M εACA and 2 mL fractions were collected. The concentration of Pg was determined at A280 using (ε1%)1cm = 1.68 as absorption coefficient (22). Each preparation was concentrated using a membrane of 10 kDa; this came to approximately 1 mg/mL using an Amicon device (Millipore). Plasminogen solutions were dialyzed overnight at 4 °C, with 0.06 M Tris, 0.06 M NaCl, 0.02 M HCl pH 8.5 (buffer A) in a dialysis tube of 25 mm.
Ion exchange chromatography
All Pgs were further purified in a column of 5 x 0.25 cm from BioRad and packed with 4 mL of DEAE Sepharose and equilibrated with buffer A. The sample was added and washed with buffer A until the A280 was ≤ 0.01 and the elution was performed by a linear gradient using buffer A and 0.07 M Tris, 0.22 M NaCl, 0.06 M HCl pH 7.5, buffer (B); 3 mL fractions were collected at a flow rate of 1.5 mL/min. The concentration of Pgs was determined and the samples were concentrated as before, then pelleted (dropping the protein solution into liquid nitrogen) and stored at -80 °C until use.
Determination of plasminogen concentration in plasma
Plasminogen concentration was determined using the Spectrolyze® Plasminogen SK kit. The determination was made in whole plasma in each step of the purification process.
Electrophoretic analysis
Gel electrophoresis was performed at denaturing (10% SDS-PAGE) conditions according to Laemmli (23). Protein samples of 5 µg were mixed with the sample buffer SDS in a 1:1 (vol/vol) ratio. Proteins were allowed to react with SDS and β-mercaptoethanol (10%) before electrophoresis and then boiled for 5 min. at 100 °C. Proteins were visualized by staining with Coomassie Brilliant Blue R. A large range of standardized markers (see materials and methods) was used.
Activation of Glu and Asp plasminogens
To 1 mg of each Pg incubatedat 37 °C, 6.72 µL of Uk were added to a final concentration of 739 IU/mL (''activated solution''). The reaction was followed spectrophotometrically at A405, using the Pm chromogenic substrate Spectrozyme as follows: To each one of eight test tubes containing 60 µl of substrate (0.3 mM), 3 µl of the activated solution were added to the substrate after 0, 1, 3, 6, 9, 15, 25 and 35 minutes of incubation; after 12 seconds the reaction with the substrate was interrupted by addition of 10 µL of 4M sodium acetate pH 3.8. The development of color determined at A405 in each test tube was recorded. The activated solution was then halted by adding 100% glycerol to a final concentration of 25% glycerol. The solution was homogenized and stored at -20 °C until use.
Determination of the plasmin concentration
According to the substrate supplier, hydrolysis of the substrate with 10 miliabsorbance (mA) (A405) at 37 °C, corresponds to 1 nM Pm. 60 µL of substrate and 3 µl of the activated solution were added to each one of three test tubes which were then incubated at 37 °C for 0, 1 and 2 min. After that, 10 µL of a ''stopping solution'' (4 M sodium acetate pH 3.8) were added and the absorbance at A405 was determined.
Protein sequence
Sequence analysis was carried out for intact Glu-Pg and degraded Lys-Pg plus the corresponding animal Pgs: 2 µg of each Pg were diluted in 500 µL of 0.1% acetic acid. Membrane pieces of 3X3 mm of polyvinilidene fluoride (PVDF) were moistened with 99.9% ethanol and then added to each acetic acid solution. The solutions were incubated at 2-8 °C for two days; they were shaken every eight hours for three minutes. The membrane pieces were washed with 20% methanol and then dried. The N-terminal sequencing was kindly performed by doctor Per-Ingvar Ohlsson at the University of Umeå, Sweden, using the Edman degradation methodology (24).
RESULTS
The three plasminogens studied displayed affinity to the Lysine-Sepharose matrix and their elution profiles from the Lysine-Sepharose column were similar. No significant contamination at washing or after elution was observed (figure n.° 1; notice, fractions 0-10 and 30-35 respectively).
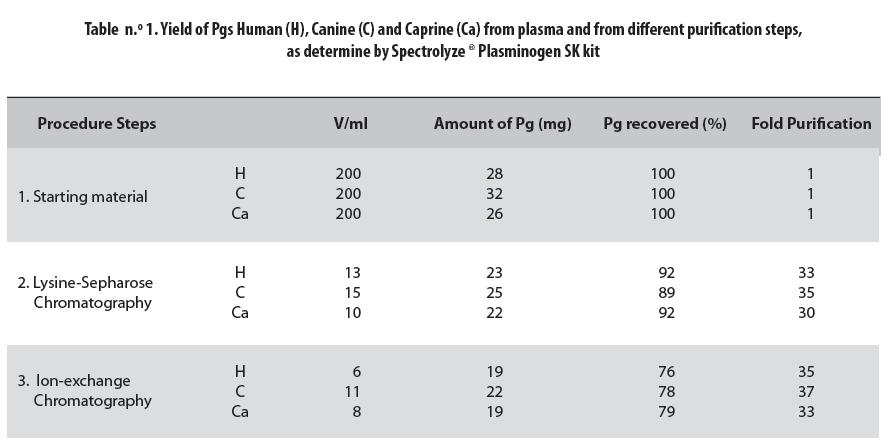
In both the starting material and the purification steps, concentration was lowest in the caprine Pg and highest in the canine one. These preparations were mixtures of Glu- and Lys-Pg in humans and Asp- and Arg-Pg, in animals.
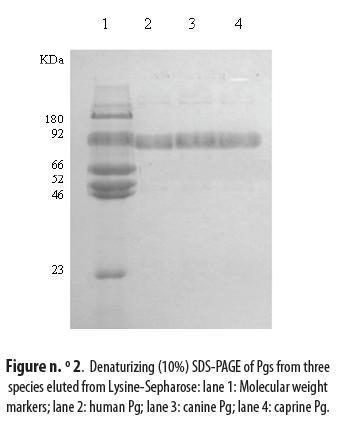
The eluted Pgs were analyzed by SDS-PAGE using a protein marker, as indicated in materials and methods, and a band of 92 kDa was identified in the three species as shown in figure n.° 2: lane 2 for human Pg, lane 3 for canine Pg and lane 4 for caprine Pg. The three bands were at the same level as the one corresponding to Glu-Pg in the marker lane 1.
Physiologically, and especially in human plasma, different types of plasminogen, such as Glu-and Lys-, have been detected. These Pgs were separated by ion exchange chromatography as shown in figure n.° 3: peak 1, not retained on the ion exchange column, was identified as intact caprine Asp-Pg, while peak 2, that was retained, corresponds to degraded caprine Arg-Pg. Plasminogens from the three species presented the same chromatogram as demonstrated in peaks 1 and 2.
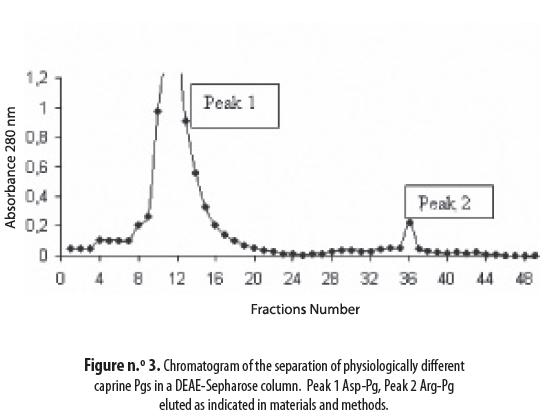
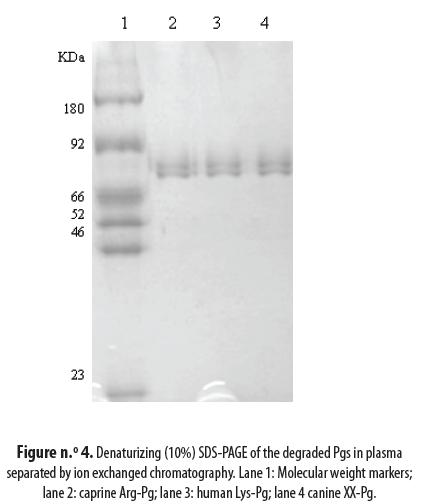
Figure n.° 4 presents the results of the electrophoretic analysis of the separation products of the different Pgs: lane 2 is the caprine Arg-Pg bound in the ion exchange chromatography column (see figure n.° 3 peak 2); lane 3 is the human Lys-Pg and lane 4 is the canine XX-Pg. The N terminal sequence was determined for both the intact and degraded Pgs. They were found to be: EPLDDY for the human Glu-Pg, XXLDDY for the canine Pg and DPLDDY for the caprine Pg. Furthermore, the human Lys-Pg N-terminal was determined to be KVYLSE; the corresponding sequences were RITLL for the caprine and RIYLS for the canine (table n.° 2).
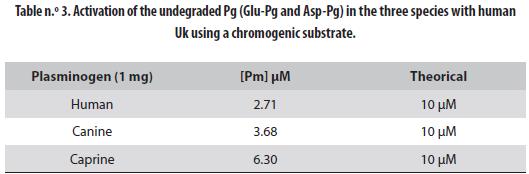
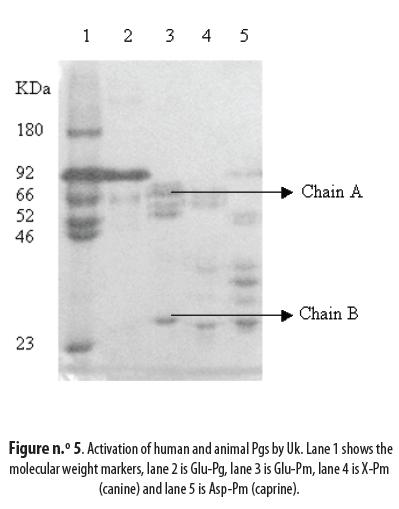
The intact Pgs, Glu- for the human and Asp- for the animal ones, were activated by Uk to Pm and the concentration of the latter in the activated solution¨ was 2.71 µM for human Glu-Pg, 3.68 µM for canine X-Pg and 6.30 µM for caprine As-Pg. These results must be compared to the maximal theoretical values of 10 µM. Human Glu-Pg presented the lowest activation, followed by the canine Pg; caprine Asp-Pg was the most activated (table n.° 3). Successful activation of the three Pgs is evidenced by the appearance of the typical A and B chains shown in figure n.° 5: lane 1 is the marker, lane 2 is Glu-Pg, lane 3 is Glu-Pm, lane 4 is canine X-Pm and lane 5 is caprine Asp-Pm.
DISCUSSION
This new purification procedure demonstrated its usefulness for plasminogens from different species. Intact human Pg and its degradation products in plasma have been purified as Glu-Pg and Lys-Pg, respectively (6); this has been confirmed in this study by the sequence determination of the two separated human Pgs. They have been used as control specimens for the corresponding animal Pgs. Human Glu- and Lys-Pgs correspond in the animals to Asp- and Arg-Pg, respectively; this has been determined from the N-terminal sequences of the intact and degraded Pgs in both human and animals. This agrees with the results of the determination of both intact Pg and degraded Pg sequence for others species (18).
This new procedure also demonstrates that Pg concentrations in plasma of the three species are similar despite the differences between species. Purification of canine Pg has been previously carried out by different methods; all of them are very complicated with more than 15 steps and no separation of the different degraded Pgs in the canine plasma (25). In this study the method was improved by using only two steps, namely: Lysine-Sepharose and ion exchange chromatograpies; furthermore, the different physiologically active animal Pgs were separated.
The first two amino-acids of the N-terminal in the intact canine Pg could not be identified. Several determinations gave the same result. No apparent reason for this fact was detected. These amino-acids can possibly be cut by Pm or other proteinases in vivo in the dog plasma. But in all animal species studied Asp has been determined as the primary amino-acid in the N-terminal sequence.
Purification of caprine Pg was performed for the first time and it displayed the same behavior as the human and canine Pgs during the purification steps. Determination of the plasma Pg concentration in the three species revealed very similar amounts. Their N-terminals had in common the LDDY sequence. The difference between human and caprine Pgs was only one amino-acid. Apparently the three species showed very similar degradation products of Pg in plasma.
Activation to Pm of the Glu-Pg and Asp-Pg by Uk generated the same bands, identified in the human case as chains A and B (26). Caprine Pg was activated to 63% (6.3 µM of 10 µM), canine Pg to 36.8% and human Pg to only 27.1%. These results indicate that the Pg/Pm system (fibrinolytic system) is activated differently in these species. In several publications from our laboratories, we have demonstrated that the Pg/Pm of the canine, equine, bovine (27), and bufalane (28) had higher affinity for substrates made for human Pg/Pm. This may indicate that the fibrinolytic system in animals probably recognizes the blood clots formed in the vasculature easier than the human system. These results agree with studies of human coagulation and fibrinolytic systems. They indicate that as a species human beings are most susceptible to thrombotic diseases (29-32). Furthermore, abnormal Pgs have been detected with different mutations that predispose patients to thrombosis (33). Relationship between Pg deficiency and different human health problems has been demonstrated (34-37).
We have no doubt that Pgs from many species can be purified by this method thus facilitating comparison of the fibrinolytic system among species and opening the possibility to identify Pgs that degrade blood clots more efficiently. Such Pgs could then be used in clinical analyzes for determination of parameters in the fibrinolytic system that cause cardiovascular problems in humans.
ACKNOWLEDGMENTS
We thank Doctor Per-Ingvar Ohlsson for performing the sequence analysis, Erasmo Meoz Hospital for supplying the human plasma samples, Doctor Carlos Mario Duque for the animal plasma specimens and the Medical Faculty at the University of Umeå in Sweden.
REFERENCES
1. Wiman B. The role of the fibrinolytic system in thrombotic disease. Acta Med Scand Suppl. 1987 Jan;Suppl.(715):169-71. [ Links ]
2. Ueshima S, Matsuo O. Development of new fibrinolytic agents. Curr Pharm Des. 2006 Jan;12(7):849-57. [ Links ]
3. Lijnen HR, Collen D. Mechanisms of physiological fibrinolysis. Baillieres Clin Haematol. 1995 Jun;8(2):277-90. [ Links ]
4. Drinane MC, Sherman JA, Hall AE, Simons M, Mulligan-Kehoe MJ. Plasminogen and plasmin activity in patients with coronary artery disease. J Thromb Haemost. 2006 Jun;4(6):1288-95. [ Links ]
5. Yamagishi K, Aleksic N, Hannan PJ, Folsom AR. Coagulation factors II, V, IX, X, XI, and XII, plasminogen, and alpha-2 antiplasmin and risk of coronary heart disease. J Atheroscler Thromb. 2010 Apr;17(4):402-9. [ Links ]
6. Wallén P, Wiman B. Characterization of human plasminogen. I. On the relationship between different molecular forms of plasminogen demonstrated in plasma and found in purified preparations. Biochim Biophys Acta. 1970 Oct;221(1):20-30. [ Links ]
7. Wiman B, Wallén P. Structural relationship between ''glutamic acid'' and ''lysine'' forms of human plasminogen and their interaction with the NH2-terminal activation peptide as studied by affinity chromatography. Eur J Biochem. 1975 Jan;50(3):489-94. [ Links ]
8. Hayes ML, Castellino FJ. Carbohydrate of the human plasminogen variants. II. Structure of the asparagine- linked oligosaccharide unit. J Biol Chem. 1979 Sep;254(18):8772-6. [ Links ]
9. Pirie-Shepherd SR, Stevens RD, Andon NL, Enghild JJ, Pizzo SV. Evidence for a novel O-linked sialylated trisaccharide on Ser-248 of human plasminogen 2. J Biol Chem. 1997 Mar;272(11):7408-11. [ Links ]
10. Robbins KC, Summaria L, Hsieh B, Shah RJ. The peptide chains of human plasmin. Mechanism of activation of human plasminogen to plasmin. J Biol Chem. 1967 May;242(10):2333-42. [ Links ]
11. Sodetz JM, Brockway WJ, Castellino FJ. Multiplicity of rabbit plasminogen. Physical characterization. Biochemistry. 1972 Nov;11(24):4451-8. [ Links ]
12. Forsgren M, Råden B, Israelsson M, Larsson K, Hedén LO. Molecular cloning and characterization of a fulllength cDNA clone for human plasminogen. FEBS Lett. 1987 Mar;213(2):254-60. [ Links ]
13. Markus G, DePasquale JL, Wissler FC. Quantitative determination of the binding of epsilonaminocaproic acid to native plasminogen. J Biol Chem. 1978 Feb;253(3):727-32. [ Links ]
14. Hoylaerts M, Lijnen HR, Collen D. Studies on the mechanism of the antifibrinolytic action of tranexamic acid. Biochim Biophys Acta. 1981 Feb;673(1):75-85. [ Links ]
15. Silverstein RL, Leung LL, Harpel PC, Nachman RL. Complex formation of platelet thrombospondin with plasminogen. Modulation of activation by tissue activator. J Clin Invest. 1984 Nov;74(5):1625-33. [ Links ]
16. Wiman B, Wallén P. The specific interaction between plasminogen and fibrin. A physiological role of the lysine binding site in plasminogen. Thromb Res. 1977 Feb;10(2):213-22. [ Links ]
17. Barrera DI, Muñoz G. A, Corredor M, Arbelaez LF. Degradación autolitíca de la plasmina e isoformas de 5 plasminogenos animales. Bistua. 2004;2(2):6-14. [ Links ]
18. Schaller J, Moser PW, Dannegger-Müller GA, Rösselet SJ, Kämpfer U, Rickli EE. Complete amino-acid sequence of bovine plasminogen. Comparison with human plasminogen. Eur J Biochem. 1985 Jun;149(2):267-78. [ Links ]
19. Paoni NF, Violand BN, Castellino FJ. Isolation and characterization of native and lower molecular weight forms of sheep plasminogen. J Biol Chem. 1977 Nov;252(21):7725-32. [ Links ]
20. Wulf RJ, Mertz ET. Studies on plasminogen. 8. Species specificity of streptokinase. Can J Biochem. 1969 Oct;47(10):927-31. [ Links ]
21. Taylor BF. Biochemistry of streptokinase. In: Markwardt F, editor(s). Fibrinolytics and Antifibrinolytics. Berlin: Springer; 1698. p. 138-149. [ Links ]
22. Deutsch DG, Mertz ET. Plasminogen: purification from human plasma by affinity chromatography. Science. 1970 Dec;170(962):1095-6. [ Links ]
23. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug;227(5259):680-5. [ Links ]
24. Edman P. Sequence determination. Mol Biol Biochem Biophys. 1970 Jan;8211-55. [ Links ]
25. Takeda Y. Plasminogen- 125 I responses in dogs to a single injection of urokinase and typhoid vaccine and to vascular injury. J Clin Invest. 1972 Jun;51(6):1363-77. [ Links ]
26. Violand BN, Castellino FJ. Mechanism of the urokinase-catalyzed activation of human plasminogen. J Biol Chem. 1976 Jul;251(13):3906-12. [ Links ]
27. Cañas Bermúdez O, Quijano Parra A, Arbeláez Ramírez LF. Cinética comparativa de la Plasmina canina con la humana, bovina y equina. Bistua. 2007;5(2):17-24. [ Links ]
28. Cañas Bermúdez O, Quijano Parra A, Arbeláez Rámirez LF. Comparación de la activación y la cinética de la plasmina bufalina con la humana. Rev Colom Cienc Pecua. 2010;23(1):47-54. [ Links ]
29. Zeng B, Bruce D, Kril J, Ploplis V, Freedman B, Brieger D. Influence of plasminogen deficiency on the contribution of polymorphonuclear leucocytes to fibrin/ogenolysis: studies in plasminogen knock-out mice. Thromb Haemost. 2002 Nov;88(5):805-10. [ Links ]
30. Busuttil SJ, Ploplis VA, Castellino FJ, Tang L, Eaton JW, Plow EF. A central role for plasminogen in the inflammatory response to biomaterials. J Thromb Haemost. 2004 Oct;2(10):1798-805. [ Links ]
31. Wang N, Zhang L, Miles L, Hoover-Plow J. Plasminogen regulates pro-opiomelanocortin processing. J Thromb Haemost. 2004 May;2(5):785-96. [ Links ]
32. Tucker HM, Simpson J, Kihiko-Ehmann M, Younkin LH, McGillis JP, Younkin SG, et al. Plasmin deficiency does not alter endogenous murine amyloid beta levels in mice. Neurosci Lett. 2004 Sep;368(3):285-9. [ Links ]
33. Aoki N, Moroi M, Sakata Y, Yoshida N, Matsuda M. Abnormal plasminogen. A hereditary molecular abnormality found in a patient with recurrent thrombosis. J Clin Invest. 1978 May;61(5):1186-95. [ Links ]
34. Mingers AM, Philapitsch A, Zeitler P, Schuster V, Schwarz HP, Kreth HW. Human homozygous type I plasminogen deficiency and ligneous conjunctivitis. APMIS. 1999 Jan;107(1):62-72. [ Links ]
35. Kraft J, Lieb W, Zeitler P, Schuster V. Ligneous conjunctivitis in a girl with severe type I plasminogen deficiency. Graefes Arch Clin Exp Ophthalmol. 2000 Sep;238(9):797-800. [ Links ]
36. Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation. 1995 Nov;92(9):2585-93. [ Links ]
37. Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995 Apr;9(7):794-807. [ Links ]
Recibido: mayo 12 de 2010
Aceptado: febrero 8 de 2011

