










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.24 no.4 Medellín Oct./Dec. 2011
PRESENTACIÓN DE CASOS
Síndrome de Cockayne: informe de dos casos clínicos y revisión de la literatura
Cockayne syndrome: report of two clinical cases and review of the literature
Cervia Margarita Palencia1; Carlos Tafur2; Víctor Jaimes3; Elizabeth Cañizalez4; Ligia Zambrano Zambrano5; Simón Gómez López6
1 Pediatra y Neuropediatra, adjunta del Servicio de Neuropediatría del Hospital de Niños Doctor Jesús García Coello, Punto Fijo, estado Falcón, Venezuela.cerviraf14@hotmail.com, cerviapalencia@hotmail.com
2 Pediatra, Puericultor y Neuropediatra, adjunto de la Unidad de Neurología Infantil del Hospital Doctor Domingo Luciani, Caracas, Venezuela.
3 Pediatra, Puericultor y Neuropediatra, adjunto de Neurología Infantil del Hospital Doctor Miguel Pérez Carreño, Caracas, Venezuela.
4 Pediatra, Puericultora y Neuropediatra, adjunta de la Unidad de Neurología Infantil del Hospital Doctor Miguel Pérez Carreño, Caracas, Venezuela.
5 Pediatra, Puericultora y Neuropediatra, adjunta de la Unidad de Neurología Infantil del Hospital Doctor Miguel Pérez Carreño. Caracas, Venezuela.
6 Pediatra, Puericultor y Neuropediatra, adjunto del Servicio de Pediatría del Hospital Luis Ortega. Porlamar, estado Nueva Esparta, Venezuela.
Recibido: agosto 06 de 2010
Aceptado: mayo 03 de 2011
RESUMEN
Introducción: el síndrome de Cockayne es un trastorno genético autosómico recesivo, caracterizado por detención del crecimiento, retraso del desarrollo, envejecimiento prematuro y fotosensibilidad. La prevalencia es de 1/100.000 nacidos vivos; es más frecuente en el sexo masculino con una relación 3:1. Desde el punto de vista genético se han descrito dos grupos: A: mutación del gen CSA (CKN1, ERCC8) en el cromosoma 5q12; B: mutación del gen CSB (ERCC6) en el cromosoma 10q11. Presentamos dos casos diagnosticados sobre bases clínicas pero en los que carecemos de estudios genéticos.
Caso 1. Niña escolar producto de padres consanguíneos quien desde el nacimiento presenta hipotonía e hipomotilidad, retardo global del desarrollo, déficit pondoestatural, cara envejecida, rasgos dismórficos, fotosensibilidad, espasticidad e hipoacusia neurosensorial y hallazgos tomográficos característicos del síndrome. Actualmente está en rehabilitación.
Caso 2. Adolescente de sexo femenino con crisis convulsivas desde los dos meses, poco progreso en el desarrollo psicomotor y pondoestatural, rasgos dismórficos y cara envejecida, hipoacusia neurosensorial bilateral, distonías repetitivas; en varias oportunidades sufrió procesos infecciosos respiratorios uno de los cuales, con neumonía bilateral, la llevó a la muerte a los 14 años.
Conclusión: se presentan estos casos y se revisa la literatura para llamar la atención sobre este síndrome de modo que se lo sospeche tempranamente en pacientes con retardo del desarrollo psicomotor, envejecimiento prematuro y fotosensibilidad. El diagnóstico temprano es la base para brindar consejería genética a los padres.
PALABRAS CLAVE
Cockayne (síndrome de), Envejecimiento Prematuro, Fotosensibilidad
SUMMARY
Introduction: Cockayne syndrome is an autosomal, recessive genetic disorder, characterized by poor growth, development impairment, premature aging, and photosensitivity. Prevalence is 1/100.000 live births, and it is more frequent in males with a ratio of 3:1. From the genetic point of view two groups have been described: Group A: mutation of the CSA gene (CKN1, ERCC8) on chromosome 5q12. Group B: mutation of the CBS gene (ERCC6) on chromosome 10q11. We report two cases that were diagnosed solely on clinical bases because no genetic studies were available.
Case 1. A school-girl, born from consanguineous parents. Since birth she has suffered from hypotonia and hypomotility. She has development delay, low weight and height gain, aged face, dysmorphic features, photosensitivity, spasticity, sensorineural hearing loss, and typical findings in the CT scan. She is currently on rehabilitation.
Case 2. A female teenager with seizures from the age of two months; she made slow progress in psychomotor development, and had low weight and height gain. Her features were dysmorphic and her face aged. She had bilateral sensorineural hearing loss, and repeated dystonias. She suffered from repeated respiratory infections and died, aged 14, from respiratory failure secondary to bilateral pneumonia.
Conclusion: We report these two cases and a review of the literature in order to attract attention to Cockayne syndrome so that early diagnoses can be made in children with psychomotor development delay, premature aging and photosensitivity. Early diagnoses are the basis for genetic counseling.
KEY WORDS
Cockayne Syndrome, Photosensitivity, Premature Aging
INTRODUCCIÓN
El síndrome de Cockayne es un trastorno genético autosómico recesivo, descrito por primera vez en 1936 por Cockayne (1). Las manifestaciones principales son detención del crecimiento, retardo del desarrollo, envejecimiento prematuro y fotosensibilidad (2-5). Es una enfermedad progresiva y de pronóstico fatal; la prevalencia es de un caso por cada 100.000 nacidos vivos (6) y es más frecuente en hombres que en mujeres: relación 3:1 (2,3,6,8).
Afecta principalmente la sustancia blanca. En la tabla 1 se resumen los criterios diagnósticos; se requiere la presencia de los criterios mayores y tres de los menores para confirmar el diagnóstico (6,7).
Clínicamente se describen tres tipos de este síndrome que difieren por su gravedad y forma de presentación: el tipo I, o forma clásica, constituye el 85% de los casos; sus manifestaciones son relativamente tardías y de evolución lenta; el tipo II es de comienzo precoz, incluso prenatal, con retardo del crecimiento intrauterino y evolución más agresiva; se llama también síndrome cerebro-óculo-facial; el tipo III, o xerodermia pigmentaria, es la forma más leve: la inteligencia es normal pero el fenotipo es característico (9-11).
Desde el punto de vista genético se han descrito dos grupos complementarios del síndrome: grupo A: mutación del gen CSA (CKN1, ERCC8) en el cromosoma 5q12, que explica el 20% de los casos; y el grupo complementario B: mutación del gen CSB (ERCC6) en el cromosoma 10q11: 80% de los casos (11-14).
Las manifestaciones clínicas incluyen: déficit pondoestatural acentuado, evidente al final del primer año de vida, que progresa hasta llevar a caquexia, microcefalia, cifoescoliosis, piel seca, pigmentada, atrófica y fotosensible que le confiere al paciente un aspecto de envejecimiento prematuro. La cara es pequeña, sumida y sin grasa; los ojos son hundidos; la nariz, pequeña y afilada; hay prognatismo y micrognatia. Desde el punto de vista ocular hay nistagmo rotatorio, catarata, degeneración retiniana, disminución de la agudeza visual, atrofia óptica y retinitis pigmentaria (1-15). Los dientes son hipoplásicos y con múltiples caries y hay sialorrea excesiva. Desde el punto de vista neurológico los pacientes presentan retardo global del desarrollo, retardo mental, sordera neurosensorial, ataxia, temblor, hipotonía que luego progresa a espasticidad, limitación de la motilidad articular y neuropatía periférica. Todos los síntomas son progresivos (2,6-8).
En algunos casos el síndrome de Cockayne se asocia a alteraciones en otros sistemas: aproximadamente el 10% de los pacientes tienen afección renal que va desde descenso en la depuración de creatinina hasta hipertensión, proteinuria y glomeruloesclerosis (9,16). Entre otras anomalías puede haber criptorquidia, mamas no desarrolladas, ciclos menstruales irregulares, hiperglicemia e hiperinsulinemia (11,17,18).
El diagnóstico es fundamentalmente clínico. Los exámenes de laboratorio pueden mostrar leucopenia, hiperlipoproteinemia, hiperglicemia e hiperinsulinemia. La tomografía computarizada (TC) muestra atrofia cortical y central con calcificación de los núcleos de la base (2,7,8,11,19). El electromiograma muestra enlentecimiento en la velocidad de conducción nerviosa (7,15,20). Los potenciales evocados auditivos confirman la sordera neurosensorial; en el estudio patológico se observan: cerebro y cerebelo pequeños, ventriculomegalia, ateroesclerosis, calcificación de los ganglios de la base, leucoencefalopatía y neuropatía periférica desmielinizante (9,11,21).
El pronóstico depende del tipo y de la evolución; la edad media de muerte es de 12 años y la mayoría de quienes lo sufren fallecen como consecuencia de neumonía u otras infecciones respiratorias (9,11,22).
En este artículo se presentan dos casos de síndrome de Cockayne tipo I, diagnosticados en nuestro servicio sobre bases clínicas pues no dispusimos de estudios genéticos, y se hace una revisión de los casos informados en la literatura, con los cuales se comparan los nuestros.
Caso 1
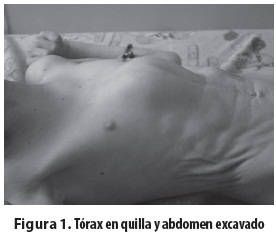
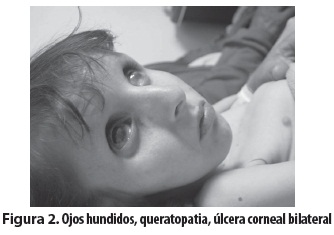
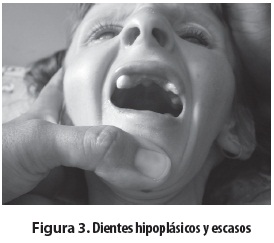
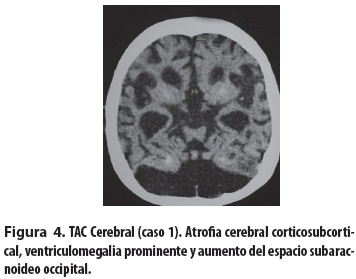
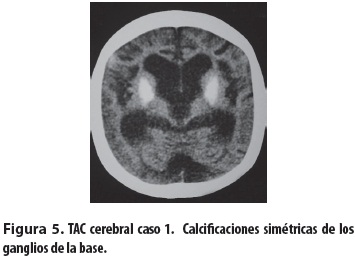
Niña escolar producto del primer embarazo, de padres consanguíneos (primos en segundo grado); embarazo complicado por toxoplasmosis tratada con espiramicina por tres meses. Parto normal, peso al nacer: 3.520 gramos, talla al nacer: 47 cm. Desde el nacimiento presentó hipotonía e hipomotricidad y posteriormente retardo del crecimiento y del desarrollo psicomotor, con mayor afectación del lenguaje y de las funciones cognitivas. Presentó sostén cefálico a los dos años y rolado a los 18 meses; solo logró la sedestación con apoyo desde los cuatro años. No ha habido desarrollo del lenguaje, solo emite sonidos guturales. Entre sus antecedentes familiares se encontró que un tío materno murió a los seis años con retardo psicomotor y dismorfias. Evaluada a los 11 años de edad se encontró lo siguiente: déficit de peso y talla (menor que el P3), perímetro cefálico: 42,7 cm (mayor que 2 DE). Piel: seca, hipotrófica, nevus y efélides menores de 5 mm y marcada fotosensibilidad. Tórax con deformidad anterior en quilla y cifoescoliosis. Abdomen: piel muy laxa, excavado (figura 1). Cara pequeña, nariz delgada aguileña, micrognatia, ojos grandes y hundidos, pabellones auriculares grandes y de implantación baja, iris azulados. Queratopatía en banda calcificada, queratitis por exposición en el ojo derecho, úlceras corneales bilaterales (figura 2); pupilas de 3 mm, buena respuesta pupilar a la luz y déficit visual, respuesta auditiva lenta, mejor al lado derecho, escasa al lado izquierdo, sialorrea, dientes hipoplásicos y faltan algunos (figura 3), dificultad para ingerir sólidos, masticación inadecuada. Hipertonía distal, postura en tijera, grado II Asworth, hipotonía axial, reflejos osteotendinosos conservados, Babinski bilateral. El hemograma, los estudios bioquímicos y de equilibrio ácido-base fueron normales. La tomografía mostró atrofia cerebral corticosubcortical, ventriculomegalia prominente y calcificaciones simétricas de los ganglios de la base (figura 4). El electroencefalograma (EEG) solo reveló lentitud, el electromiograma detectó velocidad de conducción nerviosa sensitiva y motora francamente disminuida y con los potenciales evocados auditivos del tallo cerebral (PEATC) se concluyó que había hipoacusia neurosensorial bilateral. Se le está haciendo seguimiento, con apoyo multidisciplinario y énfasis en la rehabilitación.
Caso 2
Adolescente de sexo femenino de 13 años, producto del tercer embarazo, parto normal, peso al nacer 2.800 gramos, talla al nacer 50 cm. Fue normal hasta los dos meses de edad cuando se le iniciaron crisis convulsivas parciales secundariamente generalizadas, con poco progreso en el desarrollo psicomotor y pondoestatural. Inició sedestación a los ocho meses y solo logró la bipedestación con apoyo al año de edad, sin alcanzar nunca la bipedestación independiente; tampoco hubo progreso en el lenguaje y solo alcanzó vocalización. Tiene el antecedente de un hermano fallecido al año de edad con el mismo cuadro clínico. A la exploración física se encontraron déficit pondoestatural acentuado (menor que P3), perímetro cefálico de 44 cm (menor que P25), microcefalia. Piel reseca, hipotrófica con fotosensibilidad marcada, cara pequeña con aspecto de envejecimiento, frente amplia, ojos grandes y hundidos, pterigion bilateral, pupilas mióticas, buena respuesta a la luz, úlcera corneal derecha por resequedad, no fija la mirada ni hay seguimiento visual. Simetría facial, micrognatia, dientes hipoplásicos con caries. Miembros inferiores hipotróficos, se detecta tetraparesia espástica, reflejos osteotendinosos III/IV. Hemograma, pruebas metabólicas y perfil bioquímico normales. Ecocardiograma normal. Resonancia magnética nuclear cerebral: atrofia corticosubcortical y central, agenesia del cuerpo calloso, tallo pequeño, atrofia del cerebelo, núcleos de la base calcificados bilateralmente. La electromiografía reveló disminución en la velocidad de conducción nerviosa, tanto sensitiva como motora, de predominio distal y los PEATC demostraron hipoacusia neurosensorial de predominio izquierdo. Durante su evolución persistieron las crisis convulsivas gelásticas y se le añadieron distonías repetitivas. En varias oportunidades tuvo procesos infecciosos respiratorios y murió a los 14 años por paro respiratorio secundario a neumonía bilateral.
DISCUSIÓN
El síndrome de Cockayne es de origen autosómico recesivo; en él se destacan principalmente las alteraciones neurológicas y cutáneas y el retraso del crecimiento (8). Se requiere la presencia de los criterios mayores y tres de los menores para confirmar este diagnóstico. Los dos casos que presentamos cumplían con todos los criterios mayores y más de tres de los menores (6,7). Por razones económicas no se ha podido hacer el diagnóstico genético. En algunos casos el síndrome de Cockayne se asocia a alteraciones en otros sistemas. Nuestras pacientes no las presentaron pero pudieran aparecer posteriormente en la que aún vive.
Todos los resultados de los estudios hechos a nuestras pacientes coinciden con los ya informados por otros autores: enlentecimiento de la velocidad de conducción nerviosa y alteración de los potenciales evocados auditivos del tallo cerebral (9,20,23,24,25), lentitud en el trazado electroencefalográfico (9,20), calcificaciones en los ganglios basales y atrofia cortical (9,20,23,24,25).
El pronóstico de vida es de 12 años, edad a la que murió una de nuestras pacientes debido a una enfermedad respiratoria, similar a lo descrito en la mayoría de los informes sobre este síndrome (9,11,20).
CONCLUSIÓN
El síndrome de Cockayne se debe sospechar precozmente ante un paciente con retardo del desarrollo psicomotor, envejecimiento prematuro y fotosensibilidad. Un diagnóstico oportuno permite brindar asesoría genética a los padres con miras al futuro.
REFERENCIAS BIBLIOGRÁFICAS
1. Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child. 1936 Feb;11(61):1-8. [ Links ]
2. Jones KL. Recognizable patterns of human malformation. 6th ed. Philadelphia: Elsevier Saunders; 2006. [ Links ]
3. Aicardi J. Diseases of the nervous system in childhood. London: Oxford/Blackwell editions; 1992. [ Links ]
4. Mankes JH, Sarnat HB, Bernard LM, editors. Child neurology. 5th ed. Philadelphia: Lipincott Wiliams & Wilkins; 1995. [ Links ]
5. Swaiman KF. Neurología pediátrica principios y prácticas Vol.2. 2nd ed. Mosby-Doyma libros; 1996. [ Links ]
6. Blandón B, Serrano JC. Síndrome de Cockayne: Informe de cuatro casos y revisión de la literatura. MedUNAB. 2007;10(2):133-136. [ Links ]
7. Olaciregui O, Yoldi ME, Gurtubay IG, García- Bragado F, Carrera B, Gila L, et al. [Clinical and neuropathological study of two brothers with Cockayne syndrome]. Rev Neurol. 2001;33(7):628-31. [ Links ]
8. Fejerman N, Fernández-Alvarez E, editors. Neurología pediátrica. Buenos Aires: Editorial Panamericana; 1997. [ Links ]
9. Rapin I, Weidenheim K, Lindenbaum Y, Rosenbaum P, Merchant SN, Krishna S, et al. Cockayne syndrome in adults: review with clinical and pathologic study of a new case. J Child Neurol. 2006 Nov;21(11):991-1006. [ Links ]
10. Stefanini M, Lagomarsini P, Arlett CF, Marinoni S, Borrone C, Crovato F, et al. Xeroderma pigmentosum (complementation group D) mutation is present in patients affected by trichothiodystrophy with photosensitivity. Hum Genet. 1986 Oct;74(2):107-12. [ Links ]
11. Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. 1992 Jan 1;42(1):68-84. [ Links ]
12. Hayashi M, Hayakawa K, Suzuki F, Sugita K, Satoh J, Morimatsu Y. A neuropathological study of early onset Cockayne syndrome with chromosomal anomaly 47XXX. Brain Dev. 1992 Jan;14(1):63-7. [ Links ]
13. Henning KA, Li L, Iyer N, McDaniel LD, Reagan MS, Legerski R, et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell. 1995 Aug 25;82(4):555-64. [ Links ]
14. Stefanini M, Fawcett H, Botta E, Nardo T, Lehmann AR. Genetic analysis of twenty-two patients with Cockayne syndrome. Hum Genet. 1996 Apr;97(4):418-23. [ Links ]
15. Traboulsi EI, De Becker I, Maumenee IH. Ocular findings in Cockayne syndrome. Am J Ophthalmol. 1992 Nov 15;114(5):579-83. [ Links ]
16. Komatsu A, Suzuki S, Inagaki T, Yamashita K, Hashizume K. A kindred with Cockayne syndrome caused by multiple splicing variants of the CSA gene. Am J Med Genet A. 2004 Jul 1;128A(1):67-71. [ Links ]
17. Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000 Nov 28;55(10):1442-9. [ Links ]
18. Fujimoto WY, Green ML, Seegmiller JE. Cockayne's syndrome: report of a case with hyperlipoproteinemia, hyperinsulinemia, renal disease, and normal growth hormone. J Pediatr. 1969 Nov;75(5):881-4. [ Links ]
19. Barkovich AJ. Neuroimagenología pediátrica. Buenos Aires: Ediciones Journal; 2001. [ Links ]
20. Campistol Plana J, Riverola de Veciana A, Poo Argüelles P, Colomer Oferil J, Moreno Hernández J. [Peripheral neuropathy as a presenting form of Cockayne syndrome]. Arch Neurobiol (Madr). 1991;54(4):141-5. [ Links ]
21. Soffer D, Grotsky HW, Rapin I, Suzuki K. Cockayne syndrome: unusual neuropathological findings and review of the literature. Ann Neurol. 1979 Oct;6(4):340-8. [ Links ]
22. Cao H, Williams C, Carter M, Hegele RA. CKN1 (MIM 216400): mutations in Cockayne syndrome type A and a new common polymorphism. J Hum Genet. 2004 Jan;49(1):61-3. [ Links ]
23. Arenas-Sordo M de la L, Hernández-Zamora E, Montoya-Pérez LA, Aldape-Barrios BC. Cockayne's syndrome: a case report. Literature review. Medicina oral, patología oral y cirugía bucal. 2006 May;11(3):E236-8. [ Links ]
24. Kleijer WJ, van der Sterre MLT, Garritsen VH, Raams A, Jaspers NGJ. Prenatal diagnosis of the Cockayne syndrome: survey of 15 years experience. Prenat Diagn. 2006 Oct;26(10):980-4. [ Links ]
25. Lindenbaum Y, Dickson D, Rosenbaum P, Kraemer K, Robbins I, Rapin I. Xeroderma pigmentosum/ cockayne syndrome complex: first neuropathological study and review of eight other cases. Eur J Paediatr Neurol. 2001 Jan;5(6):225-42. [ Links ]

