










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.25 no.3 Medellín July/Sept. 2012
ARTÍCULO DE REVISIÓN
Enfermedad cutánea ampollosa en el lupus eritematoso sistémico
Bullous skin disease in systemic lupus erythematosus
Luis Alonso González Naranjo1; Gloria María Vásquez Duque2; Mauricio Restrepo Escobar3
1 Profesor asistente, sección de Reumatología, Departamento de Medicina Interna, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. lagnvvn68@gmail.com
2 Profesora asociada, Sección de Reumatología, Departamento de Medicina Interna, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
3 Profesor auxiliar, Sección de Reumatología, Departamento de Medicina Interna, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
Recibido: julio 31 de 2011
Aceptado: noviembre 8 de 2011
RESUMEN
El lupus eritematoso sistémico ampolloso (LESA) es una manifestación infrecuente del lupus eritematoso sistémico (LES) causada por autoanticuerpos contra el colágeno tipo VII y otros componentes esenciales de la unión dermoepidérmica. Se presenta en pacientes que cumplen los criterios revisados de clasificación para LES del Colegio Americano de Reumatología y en presencia de alta actividad sistémica. El diagnóstico diferencial incluye otras enfermedades ampollosas autoinmunes como el penfigoide ampolloso, la epidermólisis ampollosa adquirida, la dermatosis ampollosa IgA lineal y la dermatitis herpetiforme las cuales también han sido informadas en LES.
PALABRAS CLAVE
Enfermedades Cutáneas Vesiculoampollosas, Lupus Cutáneo, Lupus Eritematoso Sistémico
SUMMARY
Bullous systemic lupus erythematosus (BSLE) is an unusual cutaneous manifestation in systemic lupus erythematosus (SLE) caused by autoantibodies targeting type VII collagen and other essential constituents of the dermal-epidermal junction. This rare entity is observed in patients who meet the American College of Rheumatology revised criteria for SLE and usually in the context of increased systemic disease activity. Differential diagnoses of BSLE include other autoimmune bullous diseases such as bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA bullous dermatosis and dermatitis herpetiformis which have also been reported in association with SLE.
KEY WORDS
Bullous Skin Diseases, Cutaneous Lupus, Systemic Lupus Erythematosus
INTRODUCCIÓN
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune, multisistémica, crónica que afecta especialmente a mujeres jóvenes durante la segunda y tercera décadas de la vida (1). Las lesiones cutáneas se presentan hasta en 90% de los pacientes (2) y son la primera manifestación de la enfermedad en 25% de los casos (3). Dichas lesiones se manifiestan en una gran variedad de formas clínicas; según la clasificación de Gilliam (4), las lesiones cutáneas del LES pueden ser específicas o inespecíficas; entre las primeras están: LE cutáneo agudo (localizado o generalizado), LE cutáneo subagudo (anular, papuloescamoso) y LE cutáneo crónico (discoide, hipertrófico o verrucoso, lupus profundus o paniculitis lúpica, lupus tumidus y lupus sabañón o perniótico). Entre las inespecíficas está el lupus eritematoso sistémico ampolloso (LESA) que es una rara enfermedad ampollosa subepidérmica mediada por anticuerpos (5-7). Esta enfermedad se caracteriza por vesículas y ampollas originadas en una base eritematosa o urticariforme semejando un penfigoide ampolloso o se puede presentar como vesículas agrupadas cuya histología es similar a la de la dermatitis herpetiforme y con características inmunológicas muy parecidas a las de la epidermólisis ampollosa adquirida (epidermolysis bullosa acquisita, EBA) (8-10).
Se reconoce el LESA como una entidad aparte asociada a autoinmunidad contra el colágeno tipo VII, un componente importante de las fibrillas de anclaje y antígeno de la EBA (11,12). Los criterios inmunopatológicos propuestos por Yell y colaboradores (13) para el LESA son similares a los de la EBA, a saber: los depósitos de inmunoglobulinas y complemento se localizan en la membrana basal en la inmunofluorescencia directa (IFD) o indirecta (IFI) y ultraestructuralmente en la lámina densa o por debajo de ella. Al igual que en la EBA, algunos pacientes con LESA tienen autoanticuerpos circulantes dirigidos contra el colágeno tipo VII (8,12); sin embargo, a pesar de la similitud inmunológica entre ambas entidades, existen algunas diferencias, a saber: el LESA afecta principalmente a pacientes jóvenes, mientras que la EBA es más frecuente en la cuarta y quinta décadas de la vida; las lesiones del LESA usualmente no dejan cicatrices, mientras que las de la EBA sí la dejan; el LESA responde a dapsona y la EBA no (8,14).
Además del colágeno tipo VII, otros componentes esenciales para la unión dermoepidérmica también son un blanco antigénico en el LESA: antígeno 1 del penfigoide ampolloso, laminina-5, laminina-6 (cadenas α3 y γ2) (15). Algunos autores consideran que el LESA es una enfermedad heterogénea, que comprende todas las enfermedades ampollosas autoinmunes en las que hay una respuesta inmune dirigida contra elementos de la membrana basal (8,16).
Epidemiología
De acuerdo con un estudio francés, la incidencia del LESA es de 0,2 casos por millón de habitantes (17). Entre 324 pacientes con enfermedades ampollosas inmunológicas adquiridas, diagnosticadas en un período de 15 años, el 1,5% tuvieron LESA (5). De un total de 67 pacientes de Singapur con enfermedades inmunoampollosas subepidérmicas, 3% tuvieron LESA, mientras que la más frecuente fue el penfigoide ampolloso (88%) (18). Menos del 5% de los pacientes con LES y manifestaciones cutáneas tienen lesiones vesiculares y ampollosas crónicas (19). El LESA afecta especialmente a adultos jóvenes entre la segunda y cuarta décadas de la vida, pero también se han informado casos en niños y en adultos mayores (5,20,21). Las mujeres, especialmente las de raza negra, son afectadas con mayor frecuencia que los hombres, aunque puede presentarse en cualquier grupo étnico. En una revisión de la literatura hecha por Lance y colaboradores (22), de 27 casos publicados, 20 (74%) fueron mujeres con edad promedio de 25 años (rango: 8 a 51 años). El predominio en mujeres jóvenes posiblemente solo refleje el patrón de distribución habitual del LES en la población (23).
Criterios diagnósticos
Los primeros criterios para el diagnóstico de LESA fueron propuestos por Camisa y Sharma en 1983 (24) (tabla 1); posteriormente, estos criterios fueron revisados aplicando técnicas de inmunofluorescencia en la piel de la lesión (25,26). Yell y colaboradores (13) revisaron estos criterios debido a la heterogeneidad de la presentación clínica e inmunohistológica de esta entidad y definieron el LESA como una enfermedad ampollosa subepidérmica adquirida en pacientes con LES, en la que los reactantes inmunes se encuentran presentes en la zona de la membrana basal en la IFD o en la IFI. Algunos de los pacientes que cumplen con los criterios para el diagnóstico de LESA tienen anticuerpos dirigidos contra el colágeno tipo VII (8,12).

Presentación clínica
El LESA se caracteriza por el comienzo agudo de un brote ampolloso generalizado, poco o nada pruriginoso (5,6), que por lo general no deja cicatriz en pacientes que cumplen con los criterios para LES establecidos por el Colegio Americano de Reumatología (27, 28). El brote puede aparecer en cualquier sitio de la piel, pero las áreas predilectas son: la zona superior del tronco, el cuello, las regiones supraclaviculares, los pliegues axilares y las superficies flexoras y extensoras de la parte proximal de las extremidades. La región malar de la cara puede estar afectada por una o múltiples vesículas pequeñas (5,6). Las áreas expuestas al sol son las más afectadas, aunque las lesiones también se pueden presentar en zonas no expuestas al sol (8). Se pueden afectar las membranas mucosas de la cavidad oral, las fosas nasales y la vulva.
Las lesiones incluyen vesículas y ampollas que surgen sobre la piel normal o eritematosa; son tensas, con líquido claro o hemorrágico y ocasionalmente se rompen dejando erosiones, costras y máculas hipopigmentadas o hiperpigmentadas; por lo general estas lesiones son múltiples, se expanden rápidamente hacia la periferia y se unen formando figuras alargadas e irregulares (figuras 1 y 2). Habitualmente no son muy pruriginosas y más bien se caracterizan por una sensación quemante. Según el predominio de las lesiones inflamatorias y la distribución de la erupción, puede imitar el penfigoide ampolloso (PA), la dermatitis herpetiforme (DH) o la variante inflamatoria de la EBA (5,8,16).
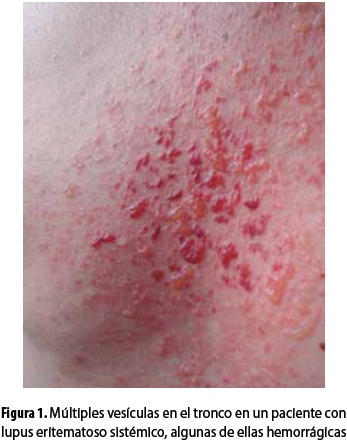
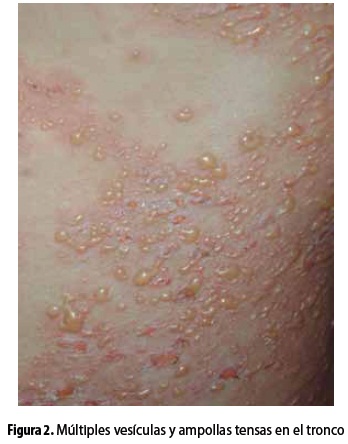
En el LESA, usualmente no se presentan las características de la variante clásica de la EBA: fragilidad cutánea, ampollas traumáticas, cicatrices y milia; sin embargo, más de una decena de casos informados de LESA se han presentado con hallazgos de EBA (15,29-34).
Frecuentemente los pacientes con LESA presentan actividad lúpica en otros órganos (10, 35-37), especialmente nefritis lúpica (26,37,38); sin embargo, el inicio y la evolución de las lesiones cutáneas pueden presentarse en ausencia de actividad lúpica en otros sistemas (8). En algunos casos el brote cutáneo ampolloso coincide con recaídas del LES, pero se ha informado su aparición entre cuatro y 12 días después del inicio de glucocorticoides sistémicos (39,40). Rara vez se presentan las lesiones ampollosas antes del diagnóstico de LES (41, 42). También se han informado casos de LESA inducidos por hidralazina (43), penicilamina (44) e interferón alfa (45).
Histopatología
Los hallazgos característicos, similares a los observados en la DH, son las ampollas subepidérmicas con acúmulos de neutrófilos que forman microabscesos en la punta de las papilas dérmicas (8,9,14,16,24,46). La coalescencia de microabscesos produce una vesícula subepidérmica tensa. La cavidad de las ampollas contiene fibrina y una gran cantidad de neutrófilos (5). La dermis se encuentra edematosa. En algunas áreas, los neutrófilos pueden estar distribuidos uniformemente en un patrón en forma de banda dentro de la dermis papilar y en la cavidad de la vesícula. También puede observarse un moderado infiltrado inflamatorio que rodea los vasos de la dermis superficial y media, el cual consiste principalmente en linfocitos, aunque también puede contener neutrófilos y eosinófilos. En ocasiones se observan vasculitis leucocitoclástica y extravasación de eritrocitos (16). La presencia de polvo nuclear que se localiza en los microabscesos papilares y en la dermis superior, en la pared de los vasos sanguíneos y alrededor de ellos, permite distinguir el LESA de otras enfermedades ampollosas como la DH, la EBA y la dermatosis ampollosa IgA lineal. Otros hallazgos histológicos que permiten diferenciar el LESA de otras enfermedades ampollosas inmunológicas son los depósitos de mucina en la dermis reticular (34) y la ausencia de eosinófilos (46).
Inmunopatología
La característica inmunopatológica del LESA es el depósito de reactantes inmunes a lo largo de la unión dermoepidérmica que se demuestra mediante IFD tanto sobre piel localizada alrededor de la lesión como sobre piel clínicamente no afectada (5). La tinción inmunofluorescente también se puede detectar en la dermis superior y ocasionalmente en los vasos de esta. Los depósitos inmunes contienen IgG, IgM e IgA: la IgG está presente en todos los casos (5,16) y la IgM, en la mitad de ellos (16); en cuanto a los depósitos de IgA, son más frecuentes en el lupus ampolloso que en el no ampolloso (76% frente a 17%) (47). Los componentes del complemento se detectan a menudo en biopsias de lesiones cutáneas y rara vez en la piel clínicamente sana (48).
El depósito de reactantes inmunes en la zona de la membrana basal tiene dos patrones: uno granular, presente en 40% de los casos, y otro lineal, comparable con la banda lúpica, en el 60% restante (16). La IFD puede ser útil para descartar la DH en la cual son más característicos los depósitos granulares de IgA y de C3 localizados en la punta de las papilas dérmicas. Si los depósitos de IgA son lineales y homogéneos se debe plantear el diagnóstico de una dermatosis ampollosa IgA lineal. La presencia concomitante de IgG e IgM en la zona de la membrana basal está a favor del diagnóstico de LESA. Sin embargo, la IFD y la IFI no distinguen entre LESA y PA en la piel intacta. Utilizando como sustrato piel separada en NaCl 1 M, se pueden observar en la IFD los depósitos de anticuerpos en el techo de la ampolla en el PA (49), mientras que en el LESA se observan en el piso o lado dérmico de la ampolla (11,50). En estudios ultraestructurales el depósito de reactantes inmunes se localiza en la lámina densa o por debajo de ella, a semejanza de lo observado en la EBA (11,14).
En pacientes con hallazgos clínicos, histológicos e inmunofluorescentes de LESA, los resultados de la IFI en piel normal son contradictorios con respecto a la presencia de anticuerpos circulantes antimembrana basal, lo cual ha llevado a establecer dos subtipos de LESA inmunológicamente diferentes, I y II, según la presencia o ausencia de anticuerpos circulantes o unidos al tejido dirigidos contra el colágeno tipo VII de la membrana basal (8).
Análisis de inmunoblot y patogénesis
Mediante el análisis de inmunoblot se ha demostrado que anticuerpos IgG antimembrana basal se unen a dos antígenos de diferente peso molecular, las proteínas de 290 y 145 kDa de la cadena alfa del colágeno tipo VII, tanto en la dermis normal como en el suero de los pacientes con LESA (5,11,51,52).
El colágeno tipo VII es el principal componente de las fibrillas de anclaje de la unión dermoepidérmica y está formado por tres cadenas alfa idénticas, cada una de ellas constituida por una estructura central helicoidal triple con colágeno de 145 kDa, una cadena larga lateral amino-terminal con dominio no colágeno de 145 kDa (NC1) y una cadena corta carboxi-terminal con dominio no colágeno de 34-kDa (NC2) (figura 3) (53).
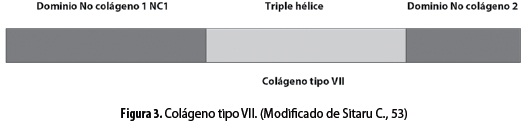
Los epítopes antigénicos principales para los autoanticuerpos en pacientes con LESA y EBA se han demostrado dentro del dominio no colágeno aminoterminal NC1 del colágeno tipo VII, justamente dentro de su región de homología a la fibronectina tipo III, la cual media la interacción entre las fibrillas de anclaje y otras proteínas de matriz (54,55).
Además de reaccionar con el NC1, el suero de pacientes con LESA y EBA también reacciona con el dominio no colágeno carboxi-terminal NC2 del colágeno tipo VII, cuando se analiza mediante las técnicas de ELISA e inmunoblot utilizando NC2 recombinante como antígeno (56).
Se han propuesto varios mecanismos para explicar la formación de ampollas por los anticuerpos contra el colágeno tipo VII tanto en el LESA como en la EBA. El primero de ellos consiste en la interferencia por parte de estos anticuerpos unidos al colágeno tipo VII con las interacciones normales entre este colágeno y sus ligandos localizados en la matriz extracelular, en la membrana basal o en la dermis papilar, lo cual debilita o bloquea las conexiones de las fibrillas de anclaje con la lámina densa o las placas de anclaje, produciendo finalmente una adherencia defectuosa entre la dermis y la lámina densa (57); el segundo mecanismo propuesto es la unión de estos anticuerpos al dominio NC2 que desestabiliza las fibrillas de anclaje al interferir con la formación de dímeros antiparalelos del colágeno tipo VII (56); el tercer mecanismo es el daño tisular inflamatorio por activación del complemento (58,59).
Mediante modelos ex vivo e in vivo en seres humanos y múridos, se ha demostrado que para la formación de vesículas son necesarias la fracción Fc de los autoanticuerpos, la activación del complemento, la liberación de gelatinasa B, elastasa y especies reactivas del oxígeno por neutrófilos activados (59).
Las funciones efectoras de la fracción Fc difieren entre los isotipos y subclases de anticuerpos. La afinidad para inhibir y activar los receptores Fc y su capacidad para activar el complemento varían de acuerdo con la subclase del anticuerpo (60). La IgG1 y la IgG3 tienen la capacidad de activar todos los tipos de receptores Fc y el complemento (59,60). Los cambios a las subclases IgG1 e IgG3 son inducidos por antígenos proteicos, mientras que la IgG4 es inducida característicamente por estimulación antigénica crónica; sin embargo, su capacidad para ejercer funciones efectoras dependientes de la fracción Fc es más débil que la de las otras subclases (61). Los carbohidratos, por su parte, provocan un cambio hacia la subclase IgG2, la cual es un activador débil del complemento (62).
Tanto en la EBA como en el LESA, los anticuerpos dirigidos contra el colágeno tipo VII son del isotipo IgG. En pacientes con EBA, los anticuerpos pertenecen a las subclases IgG1 e IgG4, mientras que en los pacientes con LESA se detectan con mayor frecuencia las subclases IgG2 e IgG3 (59,63). Recientemente se ha demostrado mediante modelos experimentales ex vivo que las subclases IgG1 e IgG3 dirigidas contra el colágeno tipo VII tienen la capacidad de activar el complemento en la unión dermoepidérmica e inducir en ella la separación dependiente del complemento y de la fracción Fc (59).
También se ha informado la presencia de otros autoanticuerpos que reaccionan con antígenos localizados en la región de la membrana basal tales como el antígeno 1 del penfigoide ampolloso y las lamiminas 5 y 6 (15). Esto puede explicarse por el fenómeno de diseminación del epítope. Este fenómeno inmunológico consiste en la liberación de epítopes antigénicos normalmente ocultos contra los cuales se genera una respuesta autoinmune secundaria luego de una lesión tisular ocasionada por un proceso autoinmune o inflamatorio primario. Por consiguiente, la reacción autoinmune primaria contra el colágeno tipo VII puede inducir una reacción inmune secundaria contra la laminina 5 y otros componentes de la membrana basal (15). La presencia de anticuerpos contra varios componentes de la zona de la membrana basal como consecuencia de la diseminación del epítope puede ser la explicación para la heterogeneidad en el fenotipo clínico y el perfil inmunológico del LESA (5).
La predisposición genética también puede ser responsable de un alto riesgo de desarrollar una respuesta autoinmune contra los antígenos de la zona de la membrana basal (64). Comparados con la población normal, tanto los pacientes con LESA como los con EBA tienen una alta incidencia del haplotipo HLA-DR2, que ha sido asociado con estados de hiperinmunidad (65).
Diagnóstico diferencial
En pacientes con LES se han informado algunas enfermedades ampollosas subepidérmicas adquiridas, tales como el penfigoide ampolloso (PA), la EBA, la dermatitis herpetiforme (DH) y la dermatosis ampollosa IgA lineal que se pueden confundir con el LESA (16,66). Su diferenciación se basa en los hallazgos clínicos, histológicos e inmunopatológicos (tabla 2) (5,16).
Tratamiento
Una característica clínica importante que diferencia el LESA de la EBA es su notable respuesta terapéutica a la dapsona (4,4-diaminodifenilsulfona) (8,14,19,26,67,68). Frecuentemente la mejoría es muy notoria: cesa la formación de nuevas ampollas en 24 a 48 horas luego de iniciado el tratamiento y se resuelven completamente las lesiones a los siete a 10 días (9,14,26,66,69). En general son efectivas las dosis bajas (25-50 mg/día), aunque a veces se requiere que sean más altas (100 mg/día) (5,26,70). Por lo general, es posible interrumpir el tratamiento con dapsona un año después de haberlo iniciado, debido a que pueden presentarse recaídas luego de su interrupción prematura. A diferencia de la DH, la disminución gradual y la posterior suspensión de la dapsona no siempre resulta en una recaída de la enfermedad (14, 52, 69). Antes de iniciar el tratamiento se recomienda medir el nivel de glucosa-6-fosfato deshidrogenasa en la sangre porque, en caso de déficit, la sulfona puede producir una anemia hemolítica grave; cabe anotar, sin embargo, que algunos pacientes han presentado anemia hemolítica durante el tratamiento con dapsona a pesar de tener niveles normales de glucosa-6-fosfato deshidrogenasa (22). Con el fin de disminuir el riesgo de hemólisis, se recomienda que la dosis de dapsona no sea mayor de 1,5 mg/kg/día (6).
Las lesiones de LESA usualmente no responden a altas dosis de glucocorticoides sistémicos (70,71), pero en algunos pacientes que no toleran la dapsona o no responden a ella se ha utilizado la combinación de altas dosis de glucocorticoides y azatioprina, aunque la resolución de las lesiones es más lenta (26,28,70,72,73) Aunque se ha utilizado con buenos resultados ciclofosfamida (28) sulfapiridina (42) y metotrexate (37), la experiencia con estas drogas es limitada. Recientemente se ha informado respuesta exitosa al tratamiento con mofetil-micofenolato (74,75) y rituximab (76) en casos refractarios.
REFERENCIAS BIBLIOGRÁFICAS
1. Rus V, Maury E, Hochberg M. The epidemiology of systemic lupus erythematosus. In: Wallace D, Hahn B, editors. Dubois' Lupus Erythematosus. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 34-44. [ Links ]
2. Petri M. Dermatologic lupus: Hopkins Lupus Cohort. Semin Cutan Med Surg. 1998 Sep;17(3):219-27. [ Links ]
3. Pistiner M, Wallace DJ, Nessim S, Metzger AL, Klinenberg JR. Lupus erythematosus in the 1980s: a survey of 570 patients. Semin Arthritis Rheum. 1991 Aug;21(1):55-64. [ Links ]
4. Gilliam JN, Sontheimer RD. Skin manifestations of SLE. Clin Rheum Dis. 1982 Apr;8(1):207-18. [ Links ]
5. Vassileva S. Bullous systemic lupus erythematosus. Clin Dermatol. 2004;22(2):129-38. [ Links ]
6. Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011 Oct;29(4):649-53. [ Links ]
7. Wallace D, Hahn B, editors. Lupus-nonspecific skin disease. In: Dubois' Lupus Erythematosus. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 621-36. [ Links ]
8. Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993 Jan;100(1):28S-34S. [ Links ]
9. Burrows NP, Bhogal BS, Black MM, Rustin MH, Ishida- Yamamoto A, Kirtschig G, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993 Mar;128(3):332-8. [ Links ]
10. Rappersberger K, Tschachler E, Tani M, Wolff K. Bullous disease in systemic lupus erythematosus. J Am Acad Dermatol. 1989 Oct;21(4 Pt 1):745-52. [ Links ]
11. Gammon WR, Woodley DT, Dole KC, Briggaman RA. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985 Jun;84(6):472-6. [ Links ]
12. Chen M, Chan LS, Cai X, O'Toole EA, Sample JC, Woodley DT. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol. 1997 Jan;108(1):68-72. [ Links ]
13. Yell JA, Allen J, Wojnarowska F, Kirtschig G, Burge SM. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995 Jun;132(6):921-8. [ Links ]
14. Hall RP, Lawley TJ, Smith HR, Katz SI. Bullous eruption of systemic lupus erythematosus. Dramatic response to dapsone therapy. Ann Intern Med. 1982 Aug;97(2):165-70. [ Links ]
15. Chan LS, Lapiere JC, Chen M, Traczyk T, Mancini AJ, Paller AS, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol. 1999 May;135(5):569-73. [ Links ]
16. Yell JA, Wojnarowska F. Bullous skin disease in lupus erythematosus. Lupus. 1997 Jan;6(2):112-21. [ Links ]
17. Bernard P, Vaillant L, Labeille B, Bedane C, Arbeille B, Denoeux JP, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol. 1995 Jan;131(1):48-52. [ Links ]
18. Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol. 2002 Sep;147(3):476-80. [ Links ]
19. Gammon W, Briggaman R. Bullous eruption of systemic lupus erythematosus. In: Wojnarowska F, Briggaman R, editors. Management of blistering diseases. London: Chapman and Hall Ltd; 1991. p. 263-75. [ Links ]
20. Tincopa M, Puttgen KB, Sule S, Cohen BA, Gerstenblith MR. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27(4):373-6. [ Links ]
21. Sáez-de-Ocariz M, Espinosa-Rosales F, López- Corella E, de León-Bojorge B. Bullous lesions as a manifestation of systemic lupus erythematosus in two Mexican teenagers. Pediatr Rheumatol Online J. 2010 Jan;8:19. [ Links ]
22. Lance N, Blaszak W, Swartz T. Bullous skin lesions in systemic lupus erythematosus. Sem Arthritis Rheum. 1991;20:396-404. [ Links ]
23. Jablonska S, Blaszczyk M. Connective tissue diseases. In: Parish L, Brenner S, Ramos-e-Silva M, editors. Atlas of women's dermatology: from infancy to maturity. New York: The Partenon Publishing Group; 2001. p. 205-217. [ Links ]
24. Camisa C, Sharma HM. Vesiculobullous systemic lupus erythematosus. Report of two cases and a review of the literature. J Am Acad Dermatol. 1983 Dec;9(6):924-33. [ Links ]
25. Camisa C, Grimwood RE. Indirect immunofluorescence in vesiculobullous eruption of systemic lupus erythematosus. J Invest Dermatol. 1986 May;86(5):606. [ Links ]
26. Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988 Jan;18(1 Pt 1):93-100. [ Links ]
27. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982 Nov;25(11):1271-7. [ Links ]
28. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997 Sep;40(9):1725. [ Links ]
29. Dotson AD, Raimer SS, Pursley TV, Tschen J. Systemic lupus erythematosus occurring in a patient with epidermolysis bullosa acquisita. Arch Dermatol. 1981 Jul;117(7):422-6. [ Links ]
30. McHenry PM, Dagg JH, Tidman MJ, Lever RS. Epidermolysis bullosa acquisita occurring in association with systemic lupus erythematosus. Clin Exp Dermatol. 1993 Jul;18(4):378-80. [ Links ]
31. Boh E, Roberts LJ, Lieu TS, Gammon WR, Sontheimer RD. Epidermolysis bullosa acquisita preceding the development of systemic lupus erythematosus. J Am Acad Dermatol. 1990 Apr;22(4):587-93. [ Links ]
32. Don PC. Vesiculobullous lupus erythematosus with milial formation. Int J Dermatol. 1992 Nov;31(11):793-5. [ Links ]
33. Prussick R, Gupta AK, Assaad DM, Hanna WM, Sauder DN. Epidermolysis bullosa acquisita with features of bullous lupus erythematosus. Int J Dermatol. 1994 Mar;33(3):192-5. [ Links ]
34. Eckman JA, Mutasim DF. Bullous systemic lupus erythematosus with milia and calcinosis. Cutis,. 2002 Jul;70(1):31-4. [ Links ]
35. Pan M, Tang H-D, Zhu H-Q, Dong J, Gill J, Zheng J. Partial epilepsy as an initial manifestation in bullous systemic lupus erythematosus. Lupus. 2011 Jul;20(8):886-90. [ Links ]
36. Miyagawa S, Shiomi Y, Fukumoto T, Nishikawa K, Ako H, Hashimoto T, et al. Bullous eruption of systemic lupus erythematosus. J Dermatol. 1994 Jun;21(6):421-5. [ Links ]
37. Malcangi G, Brandozzi G, Giangiacomi M, Zampetti M, Danieli MG. Bullous SLE: response to methotrexate and relationship with disease activity. Lupus. 2003 Jan;12(1):63-6. [ Links ]
38. Ng YY, Chang IT, Chen TW, Liou HN, Yang AH, Yang WC. Concomitant lupus nephritis and bullous eruption in systemic lupus erythematosus. Nephrol Dial Transplant. 1999 Jul;14(7):1739-43. [ Links ]
39. Lalova A, Pramatarov K, Vassileva S. Facial bullous systemic lupus erythematosus. Int J Dermatol. 1997 May;36(5):369-71. [ Links ]
40. Tsuchida T, Furue M, Kashiwado T, Ishibashi Y. Bullous systemic lupus erythematosus with cutaneous mucinosis and leukocytoclastic vasculitis. J Am Acad Dermatol. 1994 Aug;31(2 Pt 2):387-90. [ Links ]
41. Pedro SD, Dahl MV. Direct immunofluorescence of bullous systemic lupus erythematosus. Arch Dermatol. 1973 Jan;107(1):118-20. [ Links ]
42. Kettler AH, Bean SF, Duffy JO, Gammon WR. Systemic lupus erythematosus presenting as a bullous eruption in a child. Arch Dermatol. 1988 Jul;124(7):1083-7. [ Links ]
43. Fleming MG, Bergfeld WF, Tomecki KJ, Tuthill RJ, Norris M, Benedetto EA, et al. Bullous systemic lupus erythematosus. Int J Dermatol. 1989 Jun;28(5):321-6. [ Links ]
44. Condon C, Phelan M, Lyons JF. Penicillamine-induced type II bullous systemic lupus erythematosus. Br J Dermatol. 1997 Mar;136(3):474-5. [ Links ]
45. Pouthier D, Theissen F, Humbel R-L. Lupus syndrome, hypothyroidism and bullous skin lesions after interferon alfa therapy for hepatitis C in a haemodialysis patient. Nephrol Dial Transplant. 2002 Jan;17(1):174. [ Links ]
46. Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010 Aug;19(9):1050-70. [ Links ]
47. Crowson AN, Magro C. The cutaneous pathology of lupus erythematosus: a review. J Cutan Pathol. 2001 Jan;28(1):1-23. [ Links ]
48. Vassileva S. Lupus erythematosus. In: Kanitakis J, Vassileva S, Woodley D, editors. Diagnostic Immunohistochemistry of the Skin. London: Chapman & Hall; 1998. p. 144-156. [ Links ]
49. Woodley D, Sauder D, Talley MJ, Silver M, Grotendorst G, Qwarnstrom E. Localization of basement membrane components after dermalepidermal junction separation. J Invest Dermatol. 1983 Aug;81(2):149-53. [ Links ]
50. Janniger CK, Kowalewski C, Mahmood T, Lambert WC, Schwartz RA. Detection of anti-basement membrane zone antibodies in bullous systemic lupus erythematosus. J Am Acad Dermatol. 1991 Apr;24(4):643-7. [ Links ]
51. Barton DD, Fine JD, Gammon WR, Sams WM. Bullous systemic lupus erythematosus: an unusual clinical course and detectable circulating autoantibodies to the epidermolysis bullosa acquisita antigen. J Am Acad Dermatol. 1986 Aug;15(2 Pt 2):369-73. [ Links ]
52. Shirahama S, Furukawa F, Yagi H, Tanaka T, Hashimoto T, Takigawa M. Bullous systemic lupus erythematosus: detection of antibodies against noncollagenous domain of type VII collagen. J Am Acad Dermatol. 1998 May;38(5 Pt 2):844-8. [ Links ]
53. Sitaru C. Experimental models of epidermolysis bullosa acquisita. Exp Dermatol. 2007 Jun;16(6):520-31. [ Links ]
54. Jones DA, Hunt SW, Prisayanh PS, Briggaman RA, Gammon WR. Immunodominant autoepitopes of type VII collagen are short, paired peptide sequences within the fibronectin type III homology region of the noncollagenous (NC1) domain. J Invest Dermatol. 1995 Feb;104(2):231-5. [ Links ]
55. Chen M, Marinkovich MP, Jones JC, O'Toole EA, Li YY, Woodley DT. NC1 domain of type VII collagen binds to the beta3 chain of laminin 5 via a unique subdomain within the fibronectin-like repeats. J Invest Dermatol. 1999 Feb;112(2):177-83. [ Links ]
56. Chen M, Keene DR, Costa FK, Tahk SH, Woodley DT. The carboxyl terminus of type VII collagen mediates antiparallel dimer formation and constitutes a new antigenic epitope for epidermolysis Bullosa acquisita autoantibodies. J Biol Chem. 2001 Jun 15;276(24):21649-55. [ Links ]
57. Gammon WR. Hermann Pinkus Memorial Lecture--1992. Autoimmunity to collagen VII: Autoantibody-mediated pathomechanisms regulate clinical-pathological phenotypes of acquired epidermolysis bullosa and bullous SLE. J Cutan Pathol. 1993 Apr;20(2):109-14. [ Links ]
58. Gammon WR, Briggaman RA, Inman AO, Merritt CC, Wheeler CE. Evidence supporting a role for immune complex-mediated inflammation in the pathogenesis of bullous lesions of systemic lupus erythematosus. J Invest Dermatol. 1983 Oct;81(4):320-5. [ Links ]
59. Recke A, Sitaru C, Vidarsson G, Evensen M, Chiriac MT, Ludwig RJ, et al. Pathogenicity of IgG subclass autoantibodies to type VII collagen: induction of dermal-epidermal separation. J Autoimmun. 2010 Jun;34(4):435-44. [ Links ]
60. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009 Apr 16;113(16):3716-25. [ Links ]
61. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007 Sep 14;317(5844):1554-7. [ Links ]
62. Lucisano Valim YM, Lachmann PJ. The effect of antibody isotype and antigenic epitope density on the complement-fixing activity of immune complexes: a systematic study using chimaeric anti- NIP antibodies with human Fc regions. Clin Exp Immunol. 1991 Apr;84(1):1-8. [ Links ]
63. Herrero-González JE, Mascaró JM, Herrero C, Dilling A, Zillikens D, Sitaru C. Autoantibodies from patients with BSLE inducing recruitment of leukocytes to the dermoepidermal junction and subepidermal splits in cryosections of human skin. Arch Dermatol. 2006 Nov;142(11):1513-6. [ Links ]
64. Woodley DT, Sarret Y, Briggaman RA. Autoimmunity to type VII collagen. Semin Dermatol. 1991 Sep;10(3):232-9. [ Links ]
65. Gammon WR, Heise ER, Burke WA, Fine JD, Woodley DT, Briggaman RA. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. J Invest Dermatol. 1988 Sep;91(3):228-32. [ Links ]
66. Tobón GJ, Toro CE, Bravo J-C, Cañas CA. Linear IgA bullous dermatosis associated with systemic lupus erythematosus: a case report. Clin Rheumatol. 2008 Mar;27(3):391-3. [ Links ]
67. Tani M, Shimizu R, Ban M, Murata Y, Tamaki A. Systemic lupus erythematosus with vesiculobullous lesions. Immunoelectron microscopic studies. Arch Dermatol. 1984 Nov;120(11):1497-501. [ Links ]
68. Ludgate MW, Greig DE. Bullous systemic lupus erythematosus responding to dapsone. Australas J Dermatol. 2008 May;49(2):91-3. [ Links ]
69. Yung A, Oakley A. Bullous systemic lupus erythematosus. Australas J Dermatol. 2000 Nov;41(4):234-7. [ Links ]
70. Patrício P, Ferreira C, Gomes MM, Filipe P. Autoimmune bullous dermatoses: a review. Ann N Y Acad Sci. 2009 Sep;1173:203-10. [ Links ]
71. Fernández-Sánchez M, Charli-Joseph Y, Saeb- Lima M, García-Hidalgo L, Orozco-Topete R. Are corticosteroids and immunosuppressors an efficacious treatment for bullous lupus erythematosus with systemic manifestations? Eur J Dermatol. 2010;20(6):823-5. [ Links ]
72. Olansky AJ, Briggaman RA, Gammon WR, Kelly TF, Sams WM. Bullous systemic lupus erythematosus. J Am Acad Dermatol. 1982 Oct;7(4):511-20. [ Links ]
73. Jacoby RA, Abraham AA. Bullous dermatosis and systemic lupus erythematosus in a 15-year-old boy. Arch Dermatol. 1979 Sep;115(9):1094-7. [ Links ]
74. Mekouar F, Hammi S, Elomri N, Ghafir D. Bullous systemic lupus erythematosus. Intern Med. 2011 Jan;50(13):1445. [ Links ]
75. Hamminga EA, Vermeer MH. Bullous systemic lupus erythematosus responding to mycophenolate mofetil. Eur J Dermatol. 2010;20(6):844-5. [ Links ]
76. Alsanafi S, Kovarik C, Mermelstein AL, Werth VP. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011 Apr;17(3):142-4. [ Links ]

