










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.26 no.2 Medellín Apr./June 2013
PRESENTACIÓN DE CASOS
Granulomatosis con poliangitis en pediatría: a propósito de un caso con síndrome pulmón-riñón
Granulomatosis with polyangiitis in children: report of a case with kidney-lung syndrome
Carolina Muñoz Grajales1; Laura Carolina Muñoz Martínez2; Ruth María Eraso Garnica3
1 Médica Internista y Reumatóloga, Hospital Pablo Tobón Uribe y Hospital Universitario San Vicente Fundación, Medellín, Colombia. carito_mg_sp@yahoo.com
2 Médica Pediatra y Nefróloga Infantil, Medellín, Colombia.
3 Docente de Reumatología, Reumatóloga Pediatra, Universidad de Antioquia, Hospital Pablo Tobón Uribe, Medellín, Colombia.
Recibido: junio 29 de 2012
Aceptado: septiembre 6 de 2012
RESUMEN
La granulomatosis con poliangitis como causa del síndrome pulmón-riñón es infrecuente en niños. Presentamos el caso de un paciente de 13 años con hemorragia alveolar, glomerulonefritis rápidamente progresiva, escleritis y anticuerpos anti-PR3 (proteinasa 3). Hacemos referencia a las características de esta vasculitis asociada a anticuerpos contra el citoplasma de los neutrófilos (ANCA) en los niños y a su enfoque terapéutico.
PALABRAS CLAVE
Granulomatosis de Wegener, Pediatría, Vasculitis
SUMMARY
Granulomatosis with polyangiitis as a cause of kidney-lung syndrome is uncommon in children. We report the case of a 13 year old patient with alveolar hemorrhage, rapidly progressive glomerulonephritis, episcleritis and anti PR3. We refer to features of the ANCA-associated vasculitis in children and to its therapeutic approach.
KEY WORDS
Pediatrics, Vasculitis, Wegener Granulomatosis
INTRODUCCIÓN
El síndrome pulmón-riñón (SPR) caracterizado por hemorragia alveolar y glomerulonefritis obedece a variadas etiologías, con mayor frecuencia a vasculitis. Entre estas se encuentra la granulomatosis con poliangitis, que afecta vasos de pequeño y mediano calibre y se caracteriza por inflamación granulomatosa y necrosis. La granulomatosis con poliangitis (GPA) es infrecuente en la infancia, por lo que muchos de los datos disponibles provienen de estudios y series de casos en adultos. Se presenta el caso de un adolescente en el que se sospechó el diagnóstico de GPA por la presencia de síntomas constitucionales, síndrome pulmón-riñón y escleritis y se confirmó por la positividad de los ANCA (anti-neutrophil cytoplasmic antibodies) anti-PR3 y los hallazgos histológicos de glomerulonefritis necrosante pauciinmune.
PRESENTACIÓN DEL CASO
En agosto de 2011 se evaluó en el Servicio de Reumatología Infantil del Hospital Pablo Tobón Uribe a un paciente de sexo masculino de 13 años, sin antecedentes patológicos previos, que consultaba por seis meses de dolor osteomuscular, tres semanas de evolución de ojo rojo bilateral, dolor ocular y fotofobia y dos semanas de orina hiperpigmentada. Al examen físico observamos escleritis de predominio izquierdo (figura 1) y en los estudios paraclínicos se halló elevación de la proteína C reactiva (7,3 mg/dL) y de la velocidad de eritrosedimentación (87 mm/hora); asimismo, proteinuria (35 mg/m2 sc/hora), hematuria (60 eritrocitos por campo de alto poder) y elevación rápida de la creatinina (de 1,19 mg/dL a 2,2 mg/dL en cuatro días).
En las primeras 24 horas de hospitalización presentó descenso de dos gramos en la hemoglobina, sin evidencia de sangrado gastrointestinal. A pesar de la ausencia de síntomas respiratorios o hipoxemia, se decidió repetir la radiografía de tórax, que había sido normal al ingreso, para investigar la posibilidad de hemorragia alveolar. En la radiografía de control se observó un infiltrado retículo-nodular. Se solicitó tomografía axial computarizada que mostró micronódulos difusos (figura 2) y patrón de vidrio esmerilado. Con estos hallazgos se procedió a hacer lavado broncoalveolar (LBA) y biopsia de pulmón (figura 3) para descartar un proceso infeccioso o una hemorragia alveolar; se encontraron más de 50% de hemosiderófagos, directos y cultivos negativos para microorganismos y capilaritis con signos de hemorragia alveolar en la biopsia pulmonar.
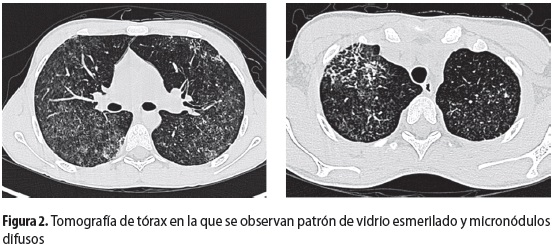
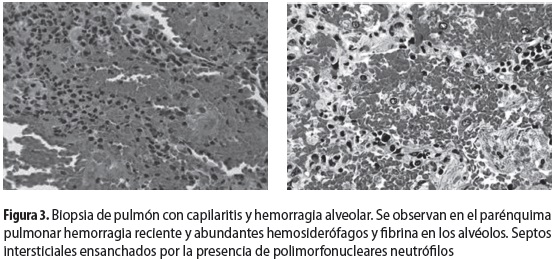
Los anticuerpos contra el citoplasma de los neutrófilos (ANCA) antiproteinasa 3 (anti-PR3), determinados por ELISA, resultaron positivos a títulos altos y la tomografía de senos paranasales descartó una afección otorrinolaringológica (ORL). Se descartaron otras causas de síndrome pulmón riñón como el lupus eritematoso sistémico y la enfermedad antimembrana basal glomerular.
Con el diagnóstico de síndrome pulmón-riñón por vasculitis asociada a ANCA, muy probablemente granulomatosis con poliangitis (GPA) se inició tratamiento con metilprednisolona endovenosa 1 gramo por tres días (para continuar con prednisolona 1 mg/kg/día por vía oral), y con ciclofosfamida endovenosa según el esquema propuesto por el Grupo Europeo de Estudio de las Vasculitis (EUVAS por la sigla en inglés de European Vasculitis Study Group) (1) (tabla 1). Sin embargo, dado que continuó con anemia progresiva sintomática y requirió diálisis por oliguria y síntomas de uremia se decidió adicionar al tratamiento el recambio plasmático (cinco sesiones) con lo que se logró el control completo de la hemorragia alveolar, pero sin recuperación de la función renal.

La biopsia renal demostró glomerulonefritis necrosante focal y segmentaria pauciinmune con proliferación extracapilar; 76% de los glomérulos con formación de medialunas, la mayoría de tipo fibrocelular, algunas fibrosas y muy pocas celulares (figura 4).
DISCUSIÓN
Se presenta el caso de un adolescente de sexo masculino en quien se sospechó el diagnóstico de granulomatosis con poliangitis (GPA) por un cuadro clínico de síntomas constitucionales, síndrome pulmón-riñón y escleritis y se confirmó por la positividad de los ANCA anti-PR3 y los hallazgos histológicos de glomerulonefritis necrosante pauciinmune.
La GPA (antes granulomatosis de Wegener) (2) es una vasculitis crónica que afecta vasos de pequeño y mediano calibre, caracterizada por inflamación granulomatosa, necrosis y presencia de anticuerpos contra el citoplasma de los neutrófilos (70% a 80%) (3,4) granulomatous sinusitis, con predilección por el tracto respiratorio tanto superior como inferior y el riñón.
Debido a la poca frecuencia de la enfermedad en niños, la mayoría de los datos disponibles provienen de estudios en adultos. Hasta donde llega nuestro conocimiento no ha habido otros reportes de casos de GPA en la literatura colombiana.
La incidencia de GPA ha aumentado tanto en adultos como en niños. En adultos ha pasado de 0,2/100.000 personas/año en los años setenta a 1,2/100.000 personas/año en los noventa (5-7); en cuanto a los niños, en un estudio canadiense reciente se observó que en los últimos cinco años la incidencia de GPA pasó de 0,28 a 0,64/100.000 personas/año (8,9).
En adultos la relación hombre/mujer es de 1,6/1 y es más frecuente entre la cuarta y sexta décadas de la vida (5,10,11).
De acuerdo con los criterios de clasificación EULAR/PRINTO/PRES (European League Against Rheumatism/Paediatric Rheumatology International Trials Organisation/Pediatric Rheumatology European Society) de 2010 (15) se considera que un paciente tiene GPA si cumple tres de los seis criterios que se presentan en la tabla 2. Nuestro paciente cumplía los criterios de clasificación, al presentar afección pulmonar, positividad de los ANCA por ELISA (anti-PR3 positivos) y afección renal definida por la hematuria y la glomerulonefritis necrosante focal y segmentaria pauciinmune; sin embargo, estos criterios al igual que los utilizados en adultos (criterios del Colegio Americano de Reumatología) no logran identificar el 100% de los pacientes con GPA ni discriminar por completo de otras vasculitis de vasos pequeños y medianos (sensibilidad y especificidad: 73,6% y 73,2%, respectivamente) (16-19) En el caso reportado, por ejemplo, en la histología pulmonar no se observaron granulomas o necrosis, pero la presencia de escleritis y los altos títulos de ANCA de tipo anti-PR3 por ELISA orientaron hacia el diagnóstico de granulomatosis con poliangitis, aunque no es posible descartar completamente la poliangitis microscópica (PAM).
Aunque la causa de las vasculitis asociadas a ANCA permanece desconocida, diferentes autores reconocen el papel de tales anticuerpos en el desarrollo de estas enfermedades.
En la técnica de inmunofluorescencia indirecta (IFI), con neutrófilos humanos fijados en etanol como sustrato, estos anticuerpos exhiben dos patrones: uno citoplasmático (c-ANCA) y uno perinuclear (p-ANCA).
El patrón de inmunofluorescencia de los ANCA suele correlacionarse, en mayor o menor grado, con el tipo de vasculitis en el que aparecen. Así, en la GPA el patrón detectado mayoritariamente es el c-ANCA y en la PAM es el p-ANCA (20-22). Mediante la prueba de ELISA (enzime-linked immunoassay), se han identificado dos tipos principales de autoanticuerpos: uno que reacciona con la proteinasa-3 (PR3-ANCA) y que produce el patrón citoplasmático (c-ANCA) en la IFI, y otro, que reacciona con la mieloperoxidasa (MPO-ANCA) y que produce el patrón perinuclear (p-ANCA) en la IFI.
Al parecer, diferentes citocinas pueden inducir un aumento de la expresión en la superficie de los neutrófilos de antígenos que reaccionan con ANCA (MPO y PR3), integrinas, receptores de quimiocinas y ligandos de selectinas, y activar el endotelio favoreciendo el incremento de la expresión de moléculas de adherencia intercelular, selectina-E y quimiocinas. Los ANCA interaccionan con sus antígenos en la superficie de los neutrófilos y se entrecruzan con los receptores Fc gamma, lo cual produce un cambio conformacional en las integrinas; posteriormente se produce adherencia por rodamiento de los neutrófilos sobre la superficie endotelial, mediada por la interacción de moléculas de adherencia intercelular y sus ligandos en el neutrófilo (integrinas); asimismo, se dan una adherencia firme y detención del movimiento del neutrófilo mediadas por la interacción entre la selectina-E y sus ligandos, por quimiocinas unidas a los glicosaminoglicanos en las células endoteliales y por receptores de quimiocinas en el neutrófilo; finalmente, los neutrófilos migran a través del endotelio, sufren explosión respiratoria y degranulación con la liberación de productos tóxicos que causan daño endotelial (23-27).
Algunos estudios han encontrado que los PR3- ANCA no solo favorecen la explosión oxidativa en neutrófilos, sino que también pueden inducir la expresión de fibras de cromatina por estas células, conocidas como NET (trampas extracelulares de los neutrófilos) (28), que en condiciones fisiológicas son útiles en el control de los procesos infecciosos, pero se pueden acumular en tejidos como el riñón e inducir inflamación, incrementar el nivel de interferón gamma (IFN-γ) y perpetuar la respuesta inflamatoria (25).
No obstante, los modelos animales para inducir la GPA utilizando anticuerpos contra PR3 no han sido exitosos, indicando que los mecanismos patogénicos en esta enfermedad no están limitados a la acción de los ANCA, sino que son más complejos (8).
En las cohortes de vasculitis sistémica en niños se observa que las manifestaciones más frecuentes al comienzo de la GPA son los síntomas generales (89%). La afección ORL se presenta en 80% de los pacientes y las manifestaciones pulmonares y renales se observan en 75% y 80%, respectivamente (13,14,29).
Las manifestaciones ORL son nasales (64,6%), sinusitis (60%) y estenosis subglótica (13,8%). Algunos autores han observado que esta última es cinco veces más común en la infancia que en los adultos (8,12,13).
En cuanto a la afección pulmonar, puede expresarse como disnea (52,5%), tos crónica (52,3%), hemoptisis/ hemorragia alveolar (44,6%) y nódulos pulmonares (42,5%); la renal se manifiesta como uroanálisis anormal (75,4%), glomerulonefritis comprobada por biopsia (52,3%) y elevación de la creatinina sérica (41,5%) (8,12-14).
Es menos frecuente la afección de los sistemas músculo-esquelético (57%), gastrointestinal (42%), nervioso (25%), de los ojos (37%) y la piel (35%). Aunque en la mayoría de las series no se ha observado afectación cardiovascular, en una cohorte de 25 pacientes se encontró trombosis venosa profunda en tres casos (12%) (8,13,14).
Aunque inicialmente algunas series sugirieron formas de la enfermedad más localizadas en la infancia, en la mayoría de las cohortes se ha observado que en los niños, como ocurrió en nuestro paciente, la GPA debuta con afectación de múltiples órganos (30,31).
Ante la ausencia de estudios en pediatría, el tratamiento de las vasculitis asociadas a ANCA en niños se extrapola del de los adultos. Antes del uso de ciclofosfamida y glucocorticoides (GC) la tasa de mortalidad a 12 meses era cercana al 80%. El 90% de los adultos logran remisión con ciclofosfamida, pero el 50% de los pacientes recaen en un período de cinco años y son frecuentes los efectos secundarios como infecciones, infertilidad y malignidad (31-34)
En 2009 se publicaron las recomendaciones del EULAR para el tratamiento de la vasculitis de pequeños y medianos vasos (tabla 3) (32); las actuales recomendaciones de tratamiento para manifestaciones graves, como las de nuestro paciente, son similares en GPA y PAM.
Es de anotar que el punto de corte de la creatinina utilizado en las recomendaciones puede no ser apropiado en los niños porque su superficie corporal y masa muscular son menores; asimismo, se debe tener presente que estas recomendaciones se basaron en la información disponible en la literatura para ese momento; los estudios más recientes con rituximab (RITUXVAS y RAVE) (33,34) no estaban disponibles.
Hasta la fecha no se han realizado ensayos clínicos en niños con GPA. En la cohorte ARChiVe, los pacientes han sido tratados con glucocorticoides (pulsos de metilprednisolona por 3-5 días seguidos de prednisolona oral) y ciclofosfamida (oral o endovenosa) para inducir la remisión, y en la mayoría de los casos con metotrexate para mantenerla (29).
En series pequeñas se han reportado otras terapias en niños, como recambio plasmático (en casos de capilaritis pulmonar o glomerulonefritis rápidamente progresiva), terapia antiagregante y rituximab en casos refractarios (35-37).
CONCLUSIONES
Se presenta el primer caso reportado en la literatura colombiana de granulomatosis con poliangitis en menores de 18 años. Dada la baja frecuencia, se requiere un alto índice de sospecha para identificar los casos de vasculitis sistémica, en particular en la población pediátrica. Pese a que la GPA no es una enfermedad frecuente en la infancia, siempre se la debe considerar ante la presencia del síndrome pulmón-riñón. El tratamiento adecuado y oportuno mejora la supervivencia de los pacientes con este tipo de vasculitis.
REFERENCIAS BIBLIOGRÁFICAS
1. de Groot K, Harper L, Jayne DRW, Flores Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009 May 19;150(10):670–80. [ Links ]
2. Falk RJ, Gross WL, Guillevin L, Hoffman GS, Jayne DRW, Jennette JC, et al. Granulomatosis with polyangiitis (Wegener's): an alternative name for Wegener's granulomatosis. Arthritis Rheum. 2011 Apr;63(4):863–4. [ Links ]
3. Holle JU, Laudien M, Gross WL. Clinical manifestations and treatment of Wegener's granulomatosis. Rheumatic Diseases Clinics of North America. 2010 Aug;36(3):507–26. [ Links ]
4. Schilder AM. Wegener's Granulomatosis vasculitis and granuloma. Autoimmun Rev. 2010 May;9(7):483–7. [ Links ]
5. Koldingsnes W, Nossent H. Epidemiology of Wegener's granulomatosis in northern Norway. Arthritis Rheum. 2000 Nov;43(11):2481–7. [ Links ]
6. Watts RA, Scott DGI, Lane SE. Epidemiology of Wegener's granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome. Cleve Clin J Med. 2002 Jan;69 Suppl 2:SII84–6. [ Links ]
7. Watts RA, Lane SE, Bentham G, Scott DG. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis Rheum. 2000 Feb;43(2):414–9. [ Links ]
8. Twilt M, Benseler S, Cabral D. Granulomatosis with polyangiitis in childhood. Curr Rheumatol Rep. 2012 Apr;14(2):107–15. [ Links ]
9. Grisaru S, Yuen GWH, Miettunen PM, Hamiwka LA. Incidence of Wegener's granulomatosis in children. J Rheumatol. 2010 Feb;37(2):440–2. [ Links ]
10. Cotch MF, Hoffman GS, Yerg DE, Kaufman GI, Targonski P, Kaslow RA. The epidemiology of Wegener's granulomatosis. Estimates of the five-year period prevalence, annual mortality, and geographic disease distribution from population-based data sources. Arthritis Rheum. 1996 Jan;39(1):87–92. [ Links ]
11. Koldingsnes W, Gran JT, Omdal R, Husby G. Wegener's granulomatosis: long-term follow-up of patients treated with pulse cyclophosphamide. Br J Rheumatol. 1998 Jun;37(6):659–64. [ Links ]
12. Rottem M, Fauci AS, Hallahan CW, Kerr GS, Lebovics R, Leavitt RY, et al. Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr. 1993 Jan;122(1):26–31. [ Links ]
13. Belostotsky VM, Shah V, Dillon MJ. Clinical features in 17 paediatric patients with Wegener granulomatosis. Pediatr Nephrol. 2002 Sep;17(9):754–61. [ Links ]
14. Akikusa JD, Schneider R, Harvey EA, Hebert D, Thorner PS, Laxer RM, et al. Clinical features and outcome of pediatric Wegener's granulomatosis. Arthritis Rheum. 2007 Jun 15;57(5):837–44. [ Links ]
15. Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 2010 May;69(5):798–806. [ Links ]
16. Fries JF, Hunder GG, Bloch DA, Michel BA, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis. Summary. Arthritis Rheum. 1990 Aug;33(8):1135–6. [ Links ]
17. Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990 Aug;33(8):1101–7. [ Links ]
18. Basu N, Watts R, Bajema I, Baslund B, Bley T, Boers M, et al. EULAR points to consider in the development of classification and diagnostic criteria in systemic vasculitis. Ann Rheum Dis. 2010 Oct;69(10):1744–50. [ Links ]
19. Watts RA, Suppiah R, Merkel PA, Luqmani R. Systemic vasculitis--is it time to reclassify? Rheumatology (Oxford). 2011 Apr;50(4):643–5. [ Links ]
20. Jennette JC, Falk RJ. Antineutrophil cytoplasmic autoantibodies and associated diseases: a review. Am J Kidney Dis. 1990 Jun;15(6):517–29. [ Links ]
21. Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med. 1988 Jun 23;318(25):1651–7. [ Links ]
22. Goldschmeding R, Dolman KM, van den Ende ME, van der Meer-Gerritsen CH, Sonnenberg A, von dem Borne AE. The relation of 29 kD C-ANCA antigen to proteinase 3. APMIS Suppl. 1990 Jan;19:26–7. [ Links ]
23. Hu N, Westra J, Kallenberg CGM. Dysregulated neutrophil--endothelial interaction in antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitides: implications for pathogenesis and disease intervention. Autoimmun Rev. 2011 Jul;10(9):536–43. [ Links ]
24. Chen M, Kallenberg CGM. New advances in the pathogenesis of ANCA-associated vasculitides. Clin Exp Rheumatol. 2009;27(1 Suppl 52):S108–14. [ Links ]
25. Kallenberg CGM. Pathophysiology of ANCA-associated small vessel vasculitis. Curr Rheumatol Rep. 2010 Dec;12(6):399–405. [ Links ]
26. Flint J, Morgan MD, Savage COS. Pathogenesis of ANCA-associated vasculitis. Rheum Dis Clin North Am. 2010 Aug;36(3):463–77. [ Links ]
27. Kallenberg CGM. Pathogenesis of PR3-ANCA associated vasculitis. J Autoimmun. 2008;30(1-2):29–36. [ Links ]
28. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009 Jun;15(6):623–5. [ Links ]
29. Cabral DA, Uribe AG, Benseler S, O'Neil KM, Hashkes PJ, Higgins G, et al. Classification, presentation, and initial treatment of Wegener's granulomatosis in childhood. Arthritis Rheum. 2009 Nov;60(11):3413–24. [ Links ]
30. Hall SL, Miller LC, Duggan E, Mauer SM, Beatty EC, Hellerstein S. Wegener granulomatosis in pediatric patients. J Pediatr. 1985 May;106(5):739–44. [ Links ]
31. Stegmayr BG, Gothefors L, Malmer B, Müller Wiefel DE, Nilsson K, Sundelin B. Wegener granulomatosis in children and young adults. A case study of ten patients. Pediatr Nephrol. 2000 Mar;14(3):208–13. [ Links ]
32. Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009 Mar;68(3):310–7. [ Links ]
33. Jones RB, Tervaert JWC, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010 Jul 15;363(3):211–20. [ Links ]
34. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010 Jul 15;363(3):221–32. [ Links ]
35. Eleftheriou D, Brogan PA. Vasculitis in children. Best Pract Res Clin Rheumatol. 2009 Jun;23(3):309–23. [ Links ]
36. Tzaribachev N, Koetter I, Kuemmerle-Deschner JB, Schedel J. Rituximab for the treatment of refractory pediatric autoimmune diseases: a case series. Cases J. 2009 Jan;2:6609. [ Links ]
37. Guerry M-JCJ, Brogan P, Bruce IN, D'Cruz DP, Harper L, Luqmani R, et al. Recommendations for the use of rituximab in anti-neutrophil cytoplasm antibodyassociated vasculitis. Rheumatology (Oxford). 2012 Apr;51(4):634–43. [ Links ]
