










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.26 no.3 Medellín July/Sept. 2013
INVESTIGACIÓN ORIGINAL
Plasmin degradation of the alpha chain of fibrinogen/fibrin: improved activation constant and activity determination in assays for tissue plasminogen activator
Degradación por la plasmina de la cadena alfa del fibrinógeno/fibrina: mejoría de la constante de activación y determinación de la actividad en ensayos para el activador del plasminógeno tisular
Tatiana M. Garcés P.1; Alfonso Quijano P.1 ; Luis Fernando Arbeláez R.1*
1 Chemistry Research Group, Health Faculty, University of Pamplona, Pamplona, Colombia.
* Chemistry Research Group, Health Faculty, University of Pamplona, Km 1 Vía Bucaramanga, Ciudadela Universitaria de Pamplona, Colombia. lui.ferar@hotmail.com; lui.ferar@hotmail.com
Recibido: enero 08 2012
Aceptado: septiembre 16 de 2012
SUMMARY
Objectives. The aim of this investigation was to increase the efficiency of ternary complex formation (fibrin-plasminogen-tissue-plasminogen activator) in the degradation process of the three-dimensional soluble fibrin monomer.
Materials and methods. Fibrinogen was purified from human plasma by repeating precipitation six times, using different concentrations of cold ethanol. Fibrinogen was converted to DesAAfibrinogen by degradation with bathroxobin. Human plasminogen was purified by affinity and ion-exchange chromatography, and activated to plasmin by incubation with urokinase. Digested DesAAfibrinogen was prepared by controlled digestion with plasmin.
Results. This study demonstrates that the α-chains of DesAAfibrinogen sterically hinder the formation of the ternary complex and are first degraded by plasmin. The degradation of fibrin(ogen) facilitates the in vitro determination of tissue plasminogen activator activity. Finally, release of fibrinopeptide A from bathroxobin-cleaved fibrinogen was confirmed, optimized and evaluated by various methods.
Conclusions. Use of digested desAAfibrinogen with plasmin yielded a more stable activation constant of the ternary complex than that of undigested DesAAfibrinogen.
KEY WORDS
Fibrin, Fibrinogen, Plasmin, Plasminogen, Tissue Plasminogen Activator
RESUMEN
Objetivos. El propósito de la presente investigación fue incrementar la eficacia de la formación del complejo terciario (fibrina-plasminógeno-activador tisular del plasminógeno) en el proceso de degradación de la estructura tridimensional del monómero de fibrina soluble.
Materiales y métodos. El fibrinógeno fue purificado de plasma humano, por seis precipitaciones repetidas, con diferentes concentraciones de etanol frío. El fibrinógeno fue convertido a desAAfibrinógeno por degradación con batroxobina. El plasminógeno humano fue purificado por cromatografías de afinidad e intercambio iónico y activado a plasmina con uroquinasa. El desAAfibrinogeno digerido fue preparado por digestión controlada con plasmina.
Resultados. Este estudio demuestra que la cadena α del desAAfibrinógeno, dificulta la formación del complejo terciario, por impedimentos estéricos, por lo cual la cadena α se sometió a hidrólisis controlada con plasmina, facilitando así la determinación in vitro de la actividad del activador tisular del plasminógeno. Finalmente, la liberación del fibrinopéptido A por hidrólisis del fibrinógeno con batroxobina, fue confirmada, optimizada y evaluada por varios métodos.
Conclusiones. El uso de desAAfibrinogeno digerido con plasmina da una constante de activación más estable en la formación del complejo terciario que el desAAfibrinógeno no digerido (fibrina-plasminogeno- activador tisular del plasminógeno).
PALABRAS CLAVE
Activador Tisular del Plasminógeno, Fibrina, Fibrinógeno, Plasmina, Plasminógeno
INTRODUCTION
Plasminogen activators (PAs) are serine proteases that convert plasminogen (plg), a widely distributed zymogen, into plasmin (pli), a trypsin-like protease of broad specificity. Two types of PAs have been identified in mammalian cells. They are produced from two related but distinct genes and are referred to as u-PA (urokinase-type) and t-PA (tissue-type) (1-3). Though they share similar catalytic activities, u-PA and t-PA differ in their molecular weights, immunological reactivities, and interactions with other proteins, such as extracellular matrix components and cell surface binding sites.
u-PA, which is produced and secreted by a variety of cell types, including monocytes/ macrophages (4, 5), trophoblasts (6,7) and epithelial cells, is thought to participate in the extracellular proteolysis that accompanies tissue remodeling and cellular invasion (1,2,8). By contrast, t-PA, which has a high affinity for fibrin (9, 10) and is synthesized by endothelial cells as a one-chain protein of 527 amino acid (a.a.) residues and 10% carbohydrate (11), constitutes an important protein in the fibrinolytic pathway, where its activity is thought to play a major role in the fibrinolytic system (12-15). The physiological role of t-PA is to activate plg to pli. The latter enzyme possesses the capability to degrade fibrin and its precursor molecule, fibrinogen (fb). The t-PA molecule is present as a single chain, and in the presence of low levels of pli (16), this protein is converted into a two-chain form, whose peptide chains are linked by a disulphide bond after hydrolysis of the peptide bond between Arg275 and Ile276. The two-chain form has similar plg activating potential as the single chain form (16).
t-PA is available for use as a thrombolytic agent (17, 18), is produced by genetic engineering (19), and is designated recombinant t-PA (rt-PA) to distinguish it from the native material.
Fibrinolysis is regulated by specific interactions between t-PA and fibrin, as well as interactions between pli and the specific pli inhibitor, α2-antiplasmin. In the t-PA assay, the fast-acting specific inhibitor for singlechain t-PA, plg activator inhibitor type 1 (PAI-1), (20, 21) is usually present in large excess over t-PA and must be blocked from quenching t-PA activity.
Abnormalities in t-PA levels are reported in a number of diseases and conditions. Patients with deep vein thrombosis are reported to have elevated PAI levels (14,15,22), and a higher risk of myocardial infarction (23) and stroke (24). In recent studies, t-PA was used as a therapeutic agent. It is effective as thrombolytic therapy, particularly for the treatment of acute ischemic stroke (25-30). Treatment with t-PA requires determination of the blood plasma t-PA concentration before and after treatment. Several assay kits are commercially available for the determination of t-PA activity and t-PA antigen concentration. In this report, we demonstrate that digested fibrin (DesAAfibrinogen) can be used to avoid the non-linear phase of the assay. Manufactures of current assay kits, particularly the t-PA activity assay, should take into account the data reported in this paper when developing a more accurate assay for determining t-PA activity.
MATERIALS AND METHODS
Chemicals
All buffer components, salts and other chemicals employed were of the highest available purity (analytical grade). Epsilon aminocaprioic acid (e-ACA), phenylmethanesulfonyl fluoride (PMSF), dimethylsulfoxide (DMSO), methanol, acetic acid and Triton X-100 were from Fluka; sodium citrate was from Merck; N,N'- methylene-bis-acrylamide, ammonium persulphate, N ,N,N',N'-tetramethylethylenediamine, β-mercaptoethanol, sodium dodecyl sulphate (SDS) were from Bio-Rad; sodium chloride (NaCl), absolute ethanol, sodium acetate and urea were from Riedel-de Haën; di-sodium hydrogen phosphate dehydrate was from Merck; Lysine-Sepharose® 4B was supplied by Amersham Biosciences; diethylaminoethyl cellulose (DEAE), Aprotinin 7 TIU/ml, and the Bicinchoninic Acid Kit for Protein Determination were supplied by Sigma; the chromogenic substrate for pli was from Spectrozyme; H-Gly-Pro-Arg-Pro-OH •2HCl, bathroxobin and t-PA were supplied by American Diagnostica Inc; and the low molecular weight marker (94 kDa (phosphorylase b), 67 kDa (bovine serum albumin), 43 kDa (ovalbumin), 30 kDa (carbonic anhydrase) 20.100 kDa (Trypsin) and 14.4 kDa (lysozyme)) was supplied by Bio-Rad.
Plasma samples
Human blood was collected in bags containing citrate, dextrose and adenine (PDA-1) as an anticoagulant, and plasma was obtained in its freshest form from the Erasmo Meoz Hospital in Cúcuta, Colombia. All samples were tested for hepatitis, VIH, Chagas and other infectious diseases, and were certified to be free of these antigens.
Human plasminogen
Affinity chromatography
Human plg was purified by affinity chromatography on Lysine-Sepharose® 4B, according to the method of Deutsch and Mertz (31), using 35 ml of Lysine-Sepharose ® 4B packed in a 12 x 2.0 cm column (Bio-Rad) and equilibrated with three column volumes of 0.1 M phosphate buffer containing 0.15 M NaCl pH 7.3 (PBS) at a flow rate of 2 ml/min. The plasma sample (200 ml) was applied and the column was washed with the same buffer until an absorbance of A280 ≤ 0.01 was achieved. Bound plg was eluted with 100 ml PBS containing 0.05M e-ACA, and 2 ml fractions were collected. The plg concentration was determined at A280 using (Ɛ1%)1cm = 1.68 as the absorption coefficient (31). The preparation was concentrated to approximately 1 mg / ml, using an Amicon device with a 10-kDa exclusion membrane (Millipore). The plg solution was dialyzed overnight at 4 °C against a buffer containing 0.06 M Tris, 0.06 M NaCl, and 0.02 M HCl pH 8.5 (Buffer A) in 25 mm dialysis tubing.
Ion exchange chromatography
Human plgs (Lys and Glu) were separated in a 5 cm x 0.25 cm column (Bio-Rad) packed with 4 ml of DEAE Sepharose FF equilibrated with buffer A. Sample was added to the column and washed with buffer A until A280 ≤ 0.01 was achieved. Elution was performed by using a linear gradient of Buffer A and 0.07 M Tris, 0.22 M NaCl, 0.06 M HCl pH 7.5 buffer (Buffer B) and a flow rate of 1.5 ml / min, according to the method of Cañas (32), and 3 ml fractions were collected.
The plg concentration was determined, and the sample was concentrated as before, converted into pellets by dropping the protein solution into liquid nitrogen, and stored at -80 °C until use.
Fibrinogen purification Fb was purified from plg-depleted plasma, according to the method used by Blömback and Blömback (33). Briefly, the plasma was precipitated six times using different concentrations of cold ethanol with constant stirring.
The precipitated fb was centrifuged and washed with ethanol after each precipitation step. The final fb preparation was resuspended in PBS buffer, and the concentration was determined at A280 using (Ɛ1%)1cm = 1.51 (33, 34) as the absorption coefficient. The fb solution was pelleted and stored at -80 °C until use.
DesAAfibrinogen
The peptide Gly-Pro-Arg-Pro-OH •2HCl was added to a final concentration of 3 g/L to 2.26 g/L fb. Bathroxobin was then added to a final concentration of 0.05 BU/ml at 25 °C. At regular time intervals (every 30 minutes) over a 5 hour period, 400 µl samples were drawn into micro-centrifuge tubes and stored at -80 °C until analysis. The assays were performed according to the method of Wiman and Rånby (35).
Isolation of fibrinopeptide A by HPLC
chromatography
DesAAfibrinogen samples of 400 µl were boiled in a water bath for 10 minutes, centrifuged for 5 minutes at 10.000 rpm, and then passed through a 0.2 µm membrane filter. A 250 µl sample was injected onto a 50 x 5 mm column of nucleophil C-18 (5 µm) equilibrated with 0.02 M ammonium acetate (adjusted to pH 6.0 with orthophosphoric acid) containing acetonitrile, 94:6 (v/v). The column was eluted over a 30 minute period at a flow rate of 1.5 ml/minute with a 0 to 50% gradient of equilibration buffer and 0.05 M ammonium acetate (adjusted to pH 6.0 with orthophosphoric acid) containing 75:25 (v/v) acetonitrile. Peptides peaks were detected at A = 210 nm and collected (36).
Determination of fibrinopeptide A concentration
Peak 2 (represents fibrinopeptide A) was collected, dialyzed against 10 mM sodium acetate pH 6.0, and concentrated in 25 mm dialysis tubing against PEG-2000.
The fibrinopeptide A concentration was determined using the Bicinchoninic Acid Kit (Sigma procedure N° BCA1) (37).
Activation of Glu-Plasminogen to Glu-Plasmin
u-PA (6.72 µl) was added to a final concentration of 739 IU/ml (''activated solution'') to 1 mg plg incubated at 37 °C. The reaction was followed spectrophotometrically at A405, using the pli chromogenic substrate, Spectrozyme. The substrate (0.3 mM; 60 µl) and 3 µl of ''activated solution'' incubated for 0, 1, 3, 6, 9, 15, 25 and 35 min were added respectively to each of eight test tubes. After 12 seconds the reaction was interrupted by adding 10 µl 4 M acetate pH 3.8. Color development was followed at A405 in each test tube. The final concentration of each test tube was then adjusted to 25% glycerol using pure glycerol, and the solution was homogenized and stored at -20 °C until use.
Determination of plasmin concentration
The hydroxylation of the substrate at 37 °C (10 mA (A405) equivalent to 1 nM pli), was performed according to the supplier's instructions.
Substrate (60 µl) and 3 µl ''activated solution'' were added to three test tubes, which were incubated at 37 °C for 0, 1 and 2 min, respectively. The reactions were stopped by adding 10 µl stopping solution (4 M acetate pH 3.8). The A405 absorbance was determined and recorded for each reaction time interval (18, 20).
Electrophoretic analysis
SDS-PAGE on a 12.5% acrylamide gel was performed under reducing conditions according to the method of Laemmli (38). Protein samples of 15 µg were mixed with SDS sample buffer (1:1 (v/v)). The samples were boiled in SDS sample buffer containing β-mercaptoethanol for 5 minutes at 100 °C and applied to the gel. Protein bands were visualized by staining the gel with Coomassie Brilliant Blue G250. A low molecular weight range marker was also applied to the gel to estimate the molecular weights of the protein bands.
Densitometry scans
Densitometry scans were performed on a General Electric scanner using the Amersham Biosciences gel scanning accessories, the gel scanning program Image Quant TL, and the software Image analysis version 2003.
Determination of t-PA activity as a function of fibrinopeptide A release
The following components were added to a 96-well micro test plate in the following order: 200 µl reagent containing 60 µg/ml Glu-plg, 0.5 mM Spectrozyme substrate for pli, 0.020 M sodium phosphate buffer (pH 7.3), 0.1 M NaCl and 1 g/L Triton X-100; 10 µl DesAAfibrinogen incubated for times ranging from 0–270 minutes; and 24 µl of t-PA (1.5 IU/ml). The micro test plate was incubated for 45 minutes at 25 °C, and the reaction was then terminated by adding 10 µl 1 M acetic acid. The absorbance at A405 was measured using a twin reader micro test plate reader (Biotek), equipped with a 25 °C incubator.
Digested DesAAfibrinogen preparation
Digested DesAAfibrinogen was prepared by controlled digestion of DesAAfibrinogen with pli. Different volumes (0, 4.6, 9.2 or 13.8 µl) of 4.3 µM pli were added to four 1-ml aliquots of DesAAfibrinogen to yield a final pli concentration of 0, 20, 40 and 60 nM, respectively. The solutions were incubated at 25 °C for 3 minutes, and the reactions were terminated by adding 8.1 µl aprotinin to a final concentration of 243 nM. A 2-µl aliquot of the digested solution was collected every 30 s and was mixed 1:1 (v/v) with electrophoresis denaturing sample buffer. The digestion was monitored electrophoretically, and the digested DesAAfibrinogen sample was stored at -80 °C until use.
Activity of t-PA with DesAAfibrinogen and digested DesAAfibrinogen
The following components were added in the following order to a micro test plate of 96 wells: 200 µl reagent containing 60 µg/ml Glu-plg, 0.5 mM Spectrozyme substrate for pli, 0.020 M sodium phosphate buffer (pH 7.3), 0.1 M NaCl and 1 g/L Triton X-100; 10 µl of DesAAfibrinogen or digested DesAAfibrinogen; 24 µl of t-PA in the activity range of 0 to 0.2 IU/ml, dissolved in the same buffer as above. All solutions were equilibrated at 37 °C, and all pipetting was performed at a constant room temperature of 37 °C. The micro test plate was incubated at 37 °C, and the difference in absorbance at 405 nm and 492 nm was determined in a twin reader micro test plate reader (Biotek) equipped with 37 °C incubator.
Determination of the activation constant
The activation constant for DesAAfibrinogen and digested DesAAfibrinogen was determined using the following equation: K = ΔA/ Δ(tPA) x t2, where ΔA is the difference in absorbance determined at 405–492 nm, Δt-PA is the difference of t-PA activity between two determination points, and t is the reaction time.
Fibrinopeptide A sequence
Sequence analysis of the HPLC peak fraction, which was dialyzed extensively in 10 mM acetic acid, was kindly performed by Doctor Per-Ingvar Ohlsson (Edman degradation methodology) at the Umeå University, Umeå, Sweden (39).
Peptide sequence
The peptide was submitted to SDS-PAGE using a 12.5% acrylamide gel. The digested DesAAfibrinogen from the SDS-PAGE gel was electroblotted to sequence grade PVDF membrane. The peptide band of 26.300 Da was cut from the membrane and processed by N-terminal amino acid sequencing (39). The peptide sequencing was kindly performed by Doctor Per-Ingvar Ohlsson (using the Edman degradation method and HPLC) at the Umeå University, Umeå, Sweden.
RESULTS
DesAAfibrinogen was prepared by allowing 0.05 BU/ml bathroxobin to convert 2.26 g/L fb to DesAAfibrinogen, in the presence of 3 g/L Gly-Pro-Arg- Pro-OH •2HCl. t-PA activity was monitored and observed to be maximal and sustained after approximately 180 minutes (Fig. 1A), and HPLC analysis confirmed that a peptide was released at a similar rate (Fig. 1B). This peptide appeared in the HPLC chromatogram at positions previously reported for fibrinopeptide A (40).
The peptide was collected during HPLC analysis, dialyzed against 10 mmol/L acetic acid, and subjected to amino acid sequence analysis. BCA protein analysis was performed before and after dialysis, and no apparent loss was observed. Amino acid sequence analysis yielded a peptide sequence that corresponded to that of fibrinopeptide A. Peaks 1 and 3 in Fig. 2 correspond to the positions of phosphorylated fibrinopeptide A (Ser-5-P) and fibrinopeptide A lacking the N-terminal alanine residue, respectively. BCA analysis of the collected peak 2 revealed that 3.75 mg fb was released about 36 µg fibrinopeptide, which corresponds to 90% of the theoretical value.
The desAAfibrinogen obtained after 180 minutes of bathroxobin digestion (Figs. 1A-B) was digested for 3 minutes by pli at final concentrations of 20, 40 or 60 nM to obtain degraded DesAAfibrinogen preparations, which were designated as digested DesAAfibrinogen 20, digested DesAAfibrinogen 40 and digested DesAA- fibrinogen 60, respectively. Fig. 3A-B shows the results of SDS-PAGE analysis of these preparations.
The SDS-PAGE gels were stained with Coomassie Brilliant Blue and submitted to densitometry analysis, (Fig. 3B). Pli digestion was seen to first affect the a-chain of DesAAfibrinogen (20 nM pli) and then the β-chain (40 nM pli).
The γ-chain appeared less sensitive to pli digestion, as can be seen in Fig. 3B, position D.
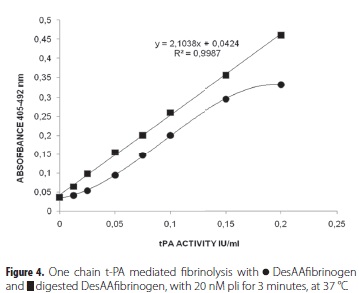
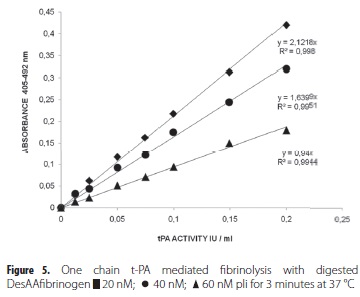
The potencies of the digested DesAAfibrinogen preparations as promoters of t-PA activity were studied, and differences in the activation constants were observed between DesAAfibrinogen and digested DesAAfibrinogen (table 1). The digested DesAAfibrinogen showed a constant and linear activation, while undigested DesAAfibrinogen showed decreasing activation. These results were confirmed by the linearity of t-PA activity in the presence of digested DesAAfibrinogen, and the low level of activity, referred to as the 'lag phase', in the presence of undigested DesAAfibrinogen (Fig. 4). Furthermore, the promotion of t-PA activity by digested DesAAfibrinogen gradually became lower when the preparations were more extensively digested with pli (60 nM) (Fig. 5). It was noted that all digested DesAAfibrinogen preparations could be used to promote t-PA activity, as evidenced by the linear standard curves.
N-terminal amino acid sequencing was performed on fibrinopeptides previously obtained by HPLC (Mw 26.300 Da) and on one peptide band from the pli digested DesAAfibrinogen preparation (Mw 26.500 Da).
The N-termini were sequenced by Edman degradation. Table 2 shows the sequence results of DesAA- fibrinogen and fibrinopeptide A, as well as the pep- tides separated by SDS-PAGE analysis after digestion of DesAAfibrinogen with pli.
DISCUSSION
The a-chain of fb is digested by bathroxobin between amino acid positions 16-17 (Arg-Gly) (41) to form DesAAfibrinogen and fibrinopeptide A. Nterminal peptide sequencing of DesAAfibrinogen (Table 2, position 1) indicated the anchor sites of bathroxobin, and that of isolated fibrinopeptide A confirmed the release of a 16 amino acid peptide with residues in the order A1-N2-S3-G4-E5.........R16 (Table 1, position 2). Upon formation, DesAAfibrinogen may act as a t-PA stimulator in t-PA assays by binding both plg and t-PA. It was noted that DesAAfibrinogen greatly enhanced the rate of plg proteolytic hydrolysis to form pli. Bathroxobin digestion of fb is highly selective at the low concentration (0.05 BU/ ml) used in the assay. HPLC analyses of digested fb showed only fibrinopeptide A, and no trace of fibrinopeptide B or any other possible peptide products (Fig. 2). Other studies show that bathroxobin is able to further digest the fb a-chain when used at higher concentrations or for longer incubation times (42). The amino acid sequence analysis confirmed that only fibrinopeptide A, with the sequence A1-N2-S3- G4-E5....R16, was released under the assay conditions used.
Digestion of DesAAfibrinogen with increasing concentrations of pli showed digestion characteristics that were different from that of bathroxobin. This was revealed by the many different peptide products seen after HPLC analysis of the products of pli digestion, even when a lower concentration of pli (20 nM) was used. Two primary sites of pli hydrolysis in the DesAAfibrinogen a-chain were identified as pli anchor sites; these were positions Lys219-Ser220, as previously described by Doolittle et al (43). (Table 2, position 3 and 4). Pli cleavage of desAAfibrinogen contributed to a more stable activation constant for the ternary complex, as compared with that of undigested DesAAfibrinogen (Table 1). This indicates a constant affinity of plg- and t-PA- for digested DesAAfibrinogen during the t-PA assay. The results of the digestion of DesAAfibrinogen with pli indicated that pli first begins its digestion of DesAAfibrinogen in the a-chain. This digestion of the a-chain occurred at especially low pli concentrations (20 nM), and within three minutes of incubation (Fig. 3b), as can be seen when the a-chain peak in position A is compared with that of position B in Fig. 3b. These results also demonstrate that it is the a-chain that interferes with DesAAfibrinogen-plg-t-PA complex formation. After digestion of DesAAfibrinogen with pli, the linearity of the t-PA assay was improved considerably when compared to the non-linearity of t-PA assays using undigested DesAAfibrinogen (Figure 4); the dissociation constant of the ternary complex with digested DesAAfibrinogen (DesAAfibrinogen-plg-t-PA) was lower than that of the ternary complex with undigested DesAAfibrinogen.
Therefore, digestion at pli concentrations greater than 20 nM is not required to produce DesAAfibrinogen or to improve the accuracy of tPA activity determinations. All these results clearly demonstrate and support the notion that the a-chain is the least important one for the binding of both plg and t-PA to DesAAfibrinogen. After degradation of DesAAfibrinogen with 60 nM pli for 3 minutes, only 8.3% of the a-chain (Fig 3b) remaining in positions A through D remained intact. Despite extensive degradation of the a-chain, the digested DesAAfibrinogen preparation still stimulated the conversion of plg to pli, and the t-PA activity determination assay was still operational (Fig. 5). These results confirm and extend the previously proposed mechanism of plg activation by t-PA in the presence of digested DesAAfibrinogen, which involves binding of both t-PA and plg.
The affinity of enzymes plg and t-PA for desAAfibrinogen dramatically increases in the presence of digested desAAfibrinogen, and at the same time, the activation constant of the reaction significantly improves, thus allowing a better determination of t-PA activity. The findings of the study presented here must be taken into account, not only in t-PA activity determinations for biological specimens such as blood plasma, but also in t-PA assays performed directly in pharmaceutical formulations and kits using chromogenic substrate, as well as for the definition of the t-PA international standard (44).
Acknowledgments
We thank Doctor Per-Ingvar Ohlsson at the University of Umeå, Umeå, Sweden, for protein sequence determinations of DesAAfibrinogen and Fibrinopeptide-A.
BIBLIOGRAPHIC REFERENCES
1. Reich E. Activation of plasminogen: a general mechanism for producing localized extracellular proteolysis. In: Berlin R, Herrmann H, Lepow I, Tanzer J, editors. Molecular basis of Biological extracellular proteolysis, Molecular Basis of Biological Degradative Processes. New York: Academic Press; 1978. p. 155–69. [ Links ]
2. Danø K, Andreasen PA, Grøndahl-Hansen J, Kristensen P, Nielsen LS, Skriver L. Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res. 1985 Jan;44:139–266. [ Links ]
3. Riccio F, Sebastio G. Human plasminogen activator Genes and protein structure. In Human Genes and Diseases,. In: F. B, editor. Human Genes and Diseases. London: John Wiley and Sons; 1986. [ Links ]
4. Unkeless JC, Gordon S, Reich E. Secretion of plasminogen activator by stimulated macrophages. J Exp Med. 1974 May 1;139(4):834–50. [ Links ]
5. Vassalli JD, Dayer JM, Wohlwend A, Belin D. Concomitant secretion of prourokinase and of a plasminogen activator- specific inhibitor by cultured human monocytesmacrophages. J Exp Med. 1984 Jul 1;159(6):1653–68. [ Links ]
6. Strickland S, Reich E, Sherman MI. Plasminogen activator in early embryogenesis: enzyme production by trophoblast and parietal endoderm. Cell. 1976 Oct;9(2):231–40. [ Links ]
7. Sappino AP, Huarte J, Belin D, Vassalli JD. Plasminogen activators in tissue remodeling and invasion: mRNA localization in mouse ovaries and implanting embryos. J Cell Biol. 1989 Dec;109(5):2471–9. [ Links ] 8. Blasi F, Vassalli JD, Danø K. Urokinase-type plasminogen activator: proenzyme, receptor, and inhibitors. J Cell Biol. 1987 May;104(4):801–4. [ Links ]
9. Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem. 1982 Mar 25;257(6):2912–9. [ Links ]
10. Suenson E, Lützen O, Thorsen S. Initial plasmin-degradation of fibrin as the basis of a positive feed-back mechanism in fibrinolysis. Eur J Biochem. 1984 May 2;140(3):513–22. [ Links ]
11. Wallén P, Pohl G, Bergsdorf N, Rånby M, Ny T, Jörnvall H. Purification and characterization of a melanoma cell plasminogen activator. Eur J Biochem. 1983 May 16;132(3):681–6. [ Links ] 12. Wiman B, Ljungberg B, Chmielewska J, Urdén G, Blombäck M, Johnsson H. The role of the fibrinolytic system in deep vein thrombosis. J Lab Clin Med. 1985 Mar;105(2):265–70. [ Links ]
13. Mellbring G, Dahlgren S, Wiman B, Sunnegårdh O. Relationship between preoperative status of the fibrinolytic system and occurrence of deep vein thrombosis after major abdominal surgery. Thromb Res. 1985 Jul 15;39(2):157–63. [ Links ]
14. Nilsson IM, Ljungnér H, Tengborn L. Two different mechanisms in patients with venous thrombosis and defective fibrinolysis: low concentration of plasminogen activator or increased concentration of plasminogen activator inhibitor. Br Med J. 1985 May 18;290(6480):1453–6. [ Links ]
15. Juhan-Vague I, Valadier J, Alessi MC, Aillaud MF, Ansaldi J, Philip-Joet C, et al. Deficient t-PA release and elevated PA inhibitor levels in patients with spontaneous or recurrent deep venous thrombosis. Thromb Haemost. 1987 Mar 3;57(1):67–72. [ Links ]
16. Rijken DC, Hoylaerts M, Collen D. Fibrinolytic properties of one-chain and two-chain human extrinsic (tissue-type) plasminogen activator. J Biol Chem. 1982 Mar 25;257(6):2920–5. [ Links ]
17. Van de Werf F, Ludbrook PA, Bergmann SR, Tiefenbrunn AJ, Fox KA, de Geest H, et al. Coronary thrombolysis with tissue-type plasminogen activator in patients with evolving myocardial infarction. N Engl J Med. 1984 Mar 8;310(10):609–13. [ Links ]
18. Bergmann SR, Fox KA, Ter-Pogossian MM, Sobel BE, Collen D. Clot-selective coronary thrombolysis with tissue-type plasminogen activator. Science. 1983 Jul 10;220(4602):1181–3. [ Links ]
19. Pennica D, Holmes WE, Kohr WJ, Harkins RN, Vehar GA, Ward CA, et al. Cloning and expression of human tissue-type plasminogen activator cDNA in E. coli. Nature. 1983 Jan 20;301(5897):214–21. [ Links ]
20. Chmielewska J, Rånby M, Wiman B. Evidence for a rapid inhibitor to tissue plasminogen activator in plasma. Thromb Res. 1983 Aug 1;31(3):427–36. [ Links ]
21. Chmielewska J, Wiman B. Determination of tissue plasminogen activator and its ''fast'' inhibitor in plasma. Clin Chem. 1986 Mar;32(3):482–5. [ Links ]
22. Wiman B, Mellbring G, Rånby M. Plasminogen activator release during venous stasis and exercise as determined by a new specific assay. Clin Chim Acta. 1983 Jan 24;127(2):279–88. [ Links ]
23. Ridker PM, Vaughan DE, Stampfer MJ, Manson JE, Hennekens CH. Endogenous tissue-type plasminogen activator and risk of myocardial infarction. Lancet. 1993 May 8;341(8854):1165–8. [ Links ]
24. Cho K-H, Lee DH, Kwon SU, Choi CG, Kim SJ, Suh D-C, et al. Factors and outcomes associated with recanalization timing after thrombolysis. Cerebrovasc Dis. 2012 Jan;33(3):255–61. [ Links ]
25. Cho K-H, Lee DH, Kwon SU, Choi CG, Kim SJ, Suh D-C, et al. Factors and outcomes associated with recanalization timing after thrombolysis. Cerebrovasc Dis. 2012 Jan;33(3):255–61. [ Links ]
26. Makihara N, Okada Y, Koga M, Shiokawa Y, Nakagawara J, Furui E, et al. Effect of serum lipid levels on stroke outcome after rt-PA therapy: SAMURAI rt-PA registry. Cerebrovasc Dis. 2012 Jan;33(3):240–7. [ Links ]
27. Jäkälä P, Jolkkonen J. Time for a neurorestorative therapy in stroke. Expert Opin Biol Ther. 2012 Mar;12(3):267–70. [ Links ]
28. Ghebre MA, Wannamethee SG, Rumley A, Whincup PH, Lowe GDO, Morris RW. Prospective study of seasonal patterns in hemostatic factors in older men and their relation to excess winter coronary heart disease deaths. J Thromb Haemost. 2012 Mar;10(3):352–8. [ Links ]
29. Barreto AD, Alexandrov A V, Lyden P, Lee J, Martin- Schild S, Shen L, et al. The argatroban and tissue-type plasminogen activator stroke study: final results of a pilot safety study. Stroke. 2012 Mar;43(3):770–5. [ Links ]
30. Ma H, Parsons MW, Christensen S, Campbell BC V, Churilov L, Connelly A, et al. A multicentre, randomized, double-blinded, placebo-controlled Phase III study to investigate EXtending the time for Thrombolysis in Emergency Neurological Deficits (EXTEND). Int J Stroke. 2012 Jan;7(1):74–80. [ Links ]
31. Deutsch DG, Mertz ET. Plasminogen: purification from human plasma by affinity chromatography. Science. 1970 Dec 4;170(3962):1095–6. [ Links ]
32. Cañas O, Arbeláez L. Activación y comparación cinética del plasminógeno equino con el plasminógeno humano. Bistúa Revista de la Facultad de Ciencias Básicas. 2007; 5: 43-52. [ Links ] 2007;5:43–52.
33. Blombäck B, Blombäck M. Purification of human and bovine fibrinogen. Arkiv för Kemi. 1956;10(9):415–43. [ Links ]
34. Vinazzer H. Basics and practice in evaluating plasminogen. Haemostasis. 1988 Jan;18 Suppl 1:41–5. [ Links ]
35. Wiman B, Rånby M. Determination of soluble fibrin in plasma by a rapid and quantitative spectrophotometric assay. Thromb Haemost. 1986 May 30;55(2):189–93. [ Links ]
36. Kehl M, Henschen A. Characterisation of the peptides released at the fibrinogen-fibrin conversion using high performance liquid chromatography. High Performance Chromatography in Protein and Peptide Chemistry,. New York: Walter de Gruyter & Co; 1981. p. 33. [ Links ]
37. Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985 Oct;150(1):76–85. [ Links ]
38. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15;227(5259):680–5. [ Links ]
39. Edman P. Sequence determination. Mol Biol Biochem Biophys. 1970 Jan;8:211–55. [ Links ]
40. Kehl M, Lottspeich F, Henschen A. High-performance liquid chromatography of proteins as applied to fibrinogen chains. Hoppe Seylers Z Physiol Chem. 1982 Dec;363(12):1501–5. [ Links ]
41. Stocker K, Fischer H, Meier J. Thrombin-like snake venom proteinases. Toxicon. 1982 Jan;20(1):265–73. [ Links ]
42. Holm B, Godall H. Degradation of fibrin by reptilase and thrombin fibrinogen and its derivatives. Ecerpta Medica International Congress No. 722. 1986. [ Links ]
43. Doolittle RF, Cassman KG, Cottrell BA, Friezner SJ, Takagi T. Amino acid sequence studies on the alpha chain of human fibrinogen. Covalent structure of the alpha-chain portion of fragment D. Biochemistry. 1977 May 19;16(8):1710–5. [ Links ]
44. Hsu J, Chiou Y, Hsu H, Chen Y, Chiou J, Lin C. Evaluation on the Analytical Methods for Tissue Plasminogen Activator (tPA) in Pharmaceutical Formulations. J Food Drug Anal. 2009;17(1):1–5. [ Links ]
