










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.26 no.3 Medellín July/Sept. 2013
PRESENTACIÓN DE CASOS
Fístula traqueoesofágica en niños: un diagnóstico para tener en cuenta. Reporte de dos casos y revisión de la literatura
Tracheo-esophageal fistula in children: a diagnosis to keep in mind. Two case reports and review of the literature
Olga Lucía Morales Múnera1; María de la Luz Valencia Chaves2; Claudia Liliana Roya Pabón3; Laura Fernanda Niño Serna4
1 Pediatra neumóloga. Profesora del Departamento de Pediatría y Puericultura, Facultad de Medicina, Universidad de Antioquia y Hospital Universitario San Vicente Fundación, Medellín, Colombia. olmmunera@yahoo.com
2 Pediatra neumóloga. Profesora del Departamento de Pediatría y Puericultura, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
3 Pediatra neumóloga. Profesora del Departamento de Pediatría y Puericultura, Facultad de Medicina, Universidad de Antioquia y Hospital Universitario San Vicente Fundación, Medellín, Colombia.
4 Residente de Pediatría, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
Recibido: julio 30 de 2012
Aceptado: septiembre 11 de 2012
RESUMEN
La fístula traqueoesofágica sin atresia esofágica es un tipo infrecuente de malformación del esófago, de etiología multifactorial, incluyendo factores ambientales y genéticos. Se manifiesta con tos y ahogo con los alimentos, cianosis y/o neumonía recurrente. El diagnóstico requiere un alto índice de sospecha clínica y su confirmación se hace por imágenes como el estudio de las vías digestivas superiores y la video-fluoroscopia, o por broncoscopia visualizando directamente la fístula, o con azul de metileno para observar el paso de este líquido. El tratamiento puede hacerse por vía endoscópica o quirúrgica, con buenos resultados.
PALABRAS CLAVE
Enfermedades de la Tráquea, Fístula Traqueoesofágica, Fluoroscopía, Tracto Gastrointestinal
SUMMARY
Tracheo-esophageal fistula without esophageal atresia is a rare type of esophageal malformation. It has a multifactorial etiology including environmental and genetic factors. Common clinical manifestations are coughing and choking after meals, cyanosis and/or recurrent pneumonia. Diagnosis requires a high clinical suspicion index. Fistula confirmation is done with imaging studies including upper digestive series, video-fluoroscopy or with the use of bronchoscopy which allows direct visualization of the fistula or methylene blue passage through the abnormal communication. Fistula closure can be done endoscopically or surgically, in both cases with good results.
KEY WORDS
Tracheoesophageal Fistula, Fluoroscopy, Gastrointestinal Tract,Tracheal Diseases
INTRODUCCIÓN
La frecuencia de malformaciones congénitas del esófa- go varía entre 1:3.000 y 1:5.000 nacidos vivos. La fístula traqueoesofágica sin atresia esofágica, o fístula en H, representa aproximadamente el 4% de estas malformaciones (1). El diagnóstico se hace generalmente en el período neonatal, pero en casos raros se puede hacer en niños mayores o en adultos. El pronóstico postoperatorio ha mejorado, aunque todavía se presentan morbilidades significativas a corto y largo plazo.
En este artículo se presentan dos casos de niños con fístula traqueoesofágica sin atresia esofágica en diferentes etapas de la vida y se hace una revisión de la literatura.
Caso 1
Niña recién nacida, producto de madre primigestante, con control prenatal adecuado. Nació de 36 semanas por cesárea debido a retardo en el crecimiento intrauterino, sin polihidramnios, con peso de 2.100 gramos y talla de 45 centímetros, no requirió reanimación cardiopulmonar. A las 13 horas de vida se inició dificultad respiratoria, evidenciada por tiraje subcostal y cianosis, que requirió reani- mación con presión positiva, por lo que fue trasladada a la unidad de cuidados intensivos neonatales con oxígeno suplementario; le iniciaron ampicilina y amikacina por sospecha de sepsis. Se hizo radiografía de tórax con resultado normal y se pasó sonda orogástrica para alimentarla sin dificultad. Siguió con taquipnea episódica, estridor y requerimiento de oxígeno. Al examen físico se evidenció tiraje subcostal y a la auscultación, crépitos. Por sospecha de neumonía se hizo nueva radiografía de tórax y se modificaron los antibióticos, iniciando cefepime (figura 1).
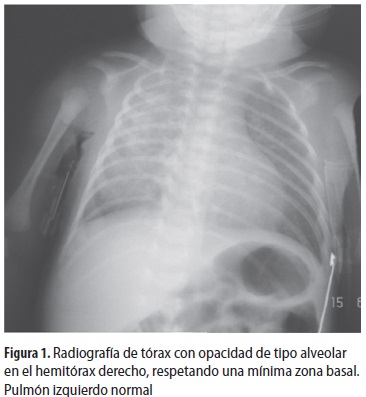
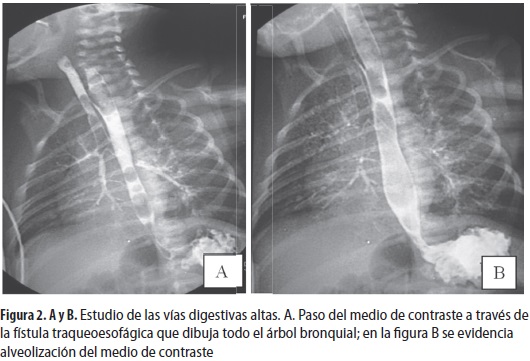
La niña siguió con dificultad respiratoria y distensión abdominal, principalmente con la alimentación, por lo que se sospechó una fístula traqueoesofágica (FTE) y se efectuó estudio de las vías digestivas altas, en el que se evidenció una fístula en esófago, a la altura de T1 que comunicaba con la tráquea a la altura de C7 en configuración de N; el medio de contraste dibujó el árbol traqueobronquial, no se observaron imágenes de reflujo ni se identificaron otros defectos (figuras 2A y 2B).
A los quince días de vida se hicieron cervicotomía y sutura de la fístula traqueoesofágica. En el ecocardiograma se hallaron comunicación interauricular, vena cava izquierda persistente y seno coronario dilatado. Los exámenes de laboratorio fueron siempre normales. A los ocho días de la cirugía se hizo un nuevo estudio de las vías digestivas altas, en el que no se evidenció paso del medio de contraste a la vía aérea.
Caso 2
Niño de 9 años, producto del segundo embarazo, pretérmino de 35 semanas sin polihidramnios, parto por cesárea ante la sospecha de hidronefrosis. Nació con ano imperforado por lo que se le hizo colostomía a las 24 horas de vida, y permaneció con ella durante cuatro meses. A los dos meses del cierre de la colostomía empezó a consultar por infección urinaria recurrente, cuyo estudio reveló un reflujo vesicoureteral grado III, que se corrigió inicialmente al año con segundo procedimiento a los 2 años.
La madre informó que desde el mes de vida se iniciaron síntomas con la alimentación caracterizados por cianosis, sialorrea escasa y tos, por lo que se hicieron estudios de las vías digestivas, que fueron normales. A los 3 meses tuvo un episodio de paro respiratorio por broncoaspiración por lo que estuvo hospitalizado en la unidad de cuidado intensivo pediátrico (UCIP) con ventilación mecánica por cinco días; se repitieron los estudios de vías digestivas con resultados normales y se hizo además pH-metría que fue patológica por lo que se inició tratamiento farmacológico. Debido a la persistencia de los síntomas de reflujo y a neumonías recurrentes, se estudiaron nuevamente las vías digestivas sin hallar alteraciones y a los 2 años de vida se hizo cirugía tipo Nissen por videolaparoscopia.
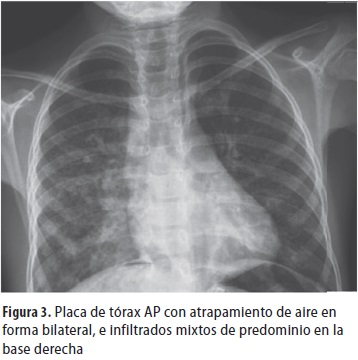
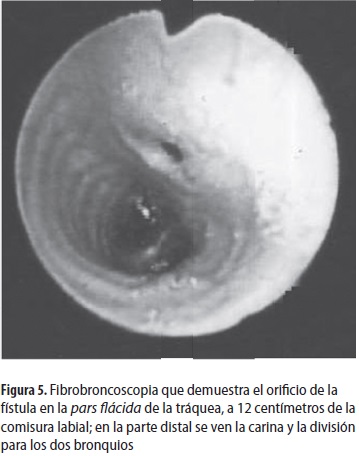
El paciente presentó posteriormente múltiples hospitalizaciones por neumonía recurrente (38 en total) y por infecciones urinarias. Los estudios para anormalidades vertebrales, cardíacas y de la inmunidad fueron normales. En su último episodio de neumonía llegó a nuestra institución donde se hallaron signos claros de neumonía; además, en el interrogatorio informaron tos con los líquidos y con la mayoría de sólidos, por lo que se hospitalizó. En la radiografía de tórax se observaron atrapamiento de aire y compromiso mixto del parénquima pulmonar principalmente en la base derecha (figura 3); se hizo videofluoroscopia por la sospecha de trastorno de la deglución o fístula traqueoesofágica; se evidenció paso del medio de contraste a la vía aérea por una fístula traqueoesofágica en H (figura 4); posteriormente se le hizo fibrobroncoscopia en la que se observó la fístula a 12 cm de la arcada dentaria con paso de azul de metileno a través de la misma (figura 5).
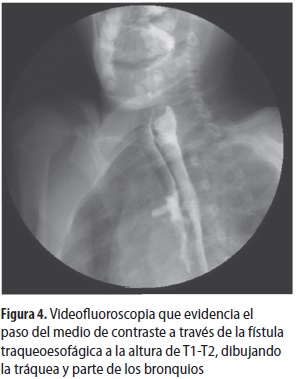
Teniendo en cuenta las alteraciones asociadas del paciente se hizo diagnóstico de asociación VACTER por ano imperforado, reflujo vesicoureteral y fístula traqueoesofágica. En junta médica con cirugía pediátrica y neumología, dadas la ubicación alta de la fístula y las posibles complicaciones del procedimiento abierto, se cerró la FTE por fibrobroncoscopia con un adhesivo hemostático de fibrina, procedimiento que se llevó a cabo sin complicaciones y con verificación del cierre, por lo que se dió de alta. En un control posterior el paciente manifestó reanudación de los síntomas con un nuevo episodio de neumonía que requirió hospitalización; se hizo nueva videofluoroscopia que mostró paso del medio de contraste a través de la FTE; en una fibrobroncoscopia se observó cierre parcial de la fístula; dado el compromiso nutricional del paciente, se hicieron gastrostomía endoscópica y posterior corrección quirúrgica de la fístula por cervicotomía sin complicaciones y con buena evolución clínica; fue dado de alta con gastrostomía.
Se hizo control al mes del procedimiento quirúrgico y, por la ganancia adecuada de peso y la ausencia de síntomas respiratorios, se retiró la gastrostomía; el paciente siguió asintomático hasta el último control, con adecuada ganancia de peso y talla con respecto a los datos pondoestaturales previos a la cirugía.
DISCUSIÓN
Las variantes de atresia esofágica (AE) y fístula traqueoesofágica (FTE) son las anomalías congénitas del esófago más frecuentes, con una tasa de prevalencia de 1/2.500 a 1/4.500 nacidos vivos, ligeramente mayor en los varones. El tipo más común es la AE proximal con FTE distal que da cuenta de 85% a 90% de los casos, seguida por la AE aislada en 5% a 8% y la FTE sin AE (también llamada fístula en H o N) en 4% (1-6); esta última, en 70% de los casos, se localiza por encima de la segunda vértebra torácica y se extiende en sentido oblicuo desde la pared posterior de la tráquea, la cual es membranosa, hasta la pared anterior del esófago (3,6,7).
No se conoce bien la etiología de estas anormalidades, pero generalmente se considera que es multifactorial. Como posibles factores de riesgo se han mencionado la diabetes mellitus gestacional, infecciones prenatales, exposición a medicamentos como antracíclicos y deficiencia de vitamina A, pero no se ha encontrado una relación contundente (4,5). La AE y la FTE se pueden presentar de forma aislada o asociadas a síndromes sin características genéticas específicas en más de 50% de los casos y con mayor incidencia en los de atresia pura (2,3,5). Entre las asociaciones más comunes están el VACTER hasta en 10%, que se caracteriza por anormalidades vertebrales, anorrectales, cardíacas, traqueoesofágicas, renales y radiales, y la asociación CHARGE compuesta por coloboma, anomalías de las orejas, atresia de las coanas, defectos cardíacos, retardo del neurodesarrollo, hipoplasia genital y alteraciones del medro. Entre los síndromes con los que la FTE está asociada frecuentemente están el de Pallister-Hall con presencia de hamartoma hipotalámico, polidactilia postaxial y central, ano imperforado y anomalías renales; el de Potter llamado también el síndrome de ''quisis'' caracterizado por labio y paladar hendidos, onfalocele e hipogenitalismo; el de Feingold que asocia alteraciones esofágicas, atresia duodenal, microcefalia, alteraciones en el aprendizaje, sindactalia y defectos cardíacos y la anemia de Fanconi (2-6,8).
En la AT, con FTE o sin ella, se han informado causas cromosómicas en 6% a 10%; entre las más frecuentes están las trisomías 13, 18 y 21. A pesar de esto, no hay datos que indiquen un papel mayor de los factores genéticos en la patogénesis de esta entidad como lo demuestran un riesgo de ocurrencia del 1% para los hermanos de estos pacientes, una tasa de concordancia entre gemelos de solo 2,5% y de ocurrencia en los hijos de un padre afectado o con la asociación VACTER de 2% a 4%, que es mucho menor en los casos aislados (2,5,6,9).
La AE/FTE se forma por una inadecuada separación del intestino primitivo anterior en las estructuras que van a formar las vías respiratorias y digestivas. Se presenta alrededor del día 22 de la gestación cuando el divertículo ventral del intestino anterior se alarga y separa la tráquea del esófago por medio de una migración cefálica de las crestas laterales del intestino anterior. El crecimiento anormal de las crestas daría origen a la AE, mientras que la falta de fusión de las mismas en la línea media daría origen a la FTE (2,3).
Se han propuesto varias teorías para que se produzca esta alteración; una de ellas plantea que hay una falla primaria del desarrollo septal a nivel de la carina; otra establece la presencia de necrosis y posterior formación de la fístula secundaria a la insuficiencia vascular localizada; una nueva teoría propone fenómenos apoptóticos como los responsables de la alteración en la separación traqueoesofágica (2,4,7).
Raramente la FTE puede ser adquirida, por lo general secundaria a tumores esofágicos, intubación prolongada, cuerpos extraños, endoprótesis, dilatación esofágica, perforaciones del esófago, divertículos, quemaduras corrosivas del esófago, abscesos mediastinales y tuberculosis. Con el tiempo ha disminuido la incidencia de esta entidad, dadas las modificaciones en la ventilación mecánica, con bajas presiones, y el manejo cuidadoso del tubo orotraqueal (10).
En los pacientes con FTE, la tráquea y el esófago son anatómicamente normales; usualmente la fístula en H va desde el nivel superior de la segunda vértebra torácica y en curso oblicuo desde la pared posterior de la tráquea a la anterior del esófago, por lo que la aspiración no ocurre en cada deglución y los líquidos tienden a pasar a través de la fístula más rápido que los sólidos; además, se considera que en el orificio se puede presentar un efecto valvular con espasmo de la musculatura alrededor que ocluye durante la deglución, dificultando y retardando así tanto el diagnóstico como el tratamiento, como fue el caso de uno de los pacientes reportados. La ausencia de epitelio ciliado en la fístula que garantizaría la limpieza de secreciones contribuye significativamente al desarrollo de neumonía recurrente en estos pacientes (2,11,12).
La forma de presentación depende del tamaño de la fístula, la edad del paciente y la relación con otras malformaciones. La sintomatología se puede manifestar desde el período neonatal, con historia de polihidramnios en los pacientes con AE pura, pero es variable ante la presencia de FTE; en estos últimos puede presentarse con crisis de cianosis, apnea, asfixia y tos durante la alimentación, con secreción constante en la vía aérea superior, síndrome de dificultad respiratoria por distensión gástrica importante, debido al aire inspirado que pasa por la fístula, elevando el diafragma y comprimiendo el pulmón como en el primer caso; el bebé puede deglutir y la sonda pasa fácilmente al estómago; además, puede tener neumonías recurrentes por aspiración, aunque la sintomatología puede ser tan sutil que no se manifieste en la edad neonatal, por lo que es importante hacer un buen interrogatorio y tener un alto índice de sospecha para que el diagnóstico no sea tardío como en el segundo de nuestros casos. En niños más grandes se han reportado casos con sintomatología recurrente o persistente (neumonías a repetición o síndrome bronco-obstructivo) como en el segundo caso reportado, o incluso pacientes asintomáticos hasta la vida adulta, debido a la presencia de una membrana cuya ruptura permite que la anomalía se haga sintomática (2,3,6,13).
Los problemas pulmonares en los pacientes con FTE se deben a la aspiración recurrente de secreciones y alimentos del esófago hacia la vía aérea, al reflujo del contenido ácido del estómago a la tráquea por la fístula y a la dismotilidad esofágica que permite la presencia de enfermedad por reflujo gastroesofágico; cuando el diagnóstico es tardío, por lo general hay afectación nutricional (3).
En los pacientes con FTE los volúmenes aspirados pueden ser grandes o masivos con sintomatología aguda, como lo que ocurrió en nuestro primer caso, o pueden ser pequeños que llevan a inflamación permanente de la vía aérea y del parénquima pulmonar dando lugar a neumonías a repetición como las de nuestro segundo caso. Algunos pacientes pueden presentar bronquiectasias o bronquiolitis obliterante (2,3).
Para llegar al diagnóstico de FTE con AE o sin ella, se requiere inicialmente una buena historia clínica, además de la alta sospecha generada en los estudios ecográficos prenatales de pacientes con polihidramnios que pueden revelar un área anecoica en el centro del cuello, ausencia de burbuja gástrica, dilatación de la papila ciega que termina en forma de bolsa a nivel del esófago, que en el caso de la atresia pura de esófago tiene un valor predictivo positivo de 75% a 91% y cuando se acompaña de FTE va de 44% a 56%; también puede haber alteraciones en la resonancia fetal, que obligan a hacer un estudio ecocardiográfico y amniocentesis prenatal y además sirven para planear el parto en una institución que atienda este tipo de condiciones (2,14,15). En el examen del neonato se debe tratar de pasar una sonda orogástrica: si no pasa, apoya el diagnóstico de AE (2).
La radiografía de tórax puede evidenciar ausencia de la radiolucidez gástrica, hipoplasia pulmonar que se puede presentar por el escape de líquido amniótico al tracto gastrointestinal a través de la fístula y, en casos de diagnóstico tardío, neumonía recurrente (2,3,14). El diagnóstico certero de FTE se establece con el estudio de las vías digestivas superiores que idealmente debe hacerse con un medio hidrosoluble. Para el caso de la FTE en H, tal estudio tiene una sensibilidad de 50% a 73%, identificando el paso del medio de contraste hacia la vía aérea. La videofluoroscopia también permite evidenciar dicha anormalidad. Cuando es difícil demostrar su presencia como en los casos de fístula en H, se puede poner al paciente en posición prona y administrar el medio de contraste a través de una sonda en el esófago distal que lentamente se va retirando, lo que puede ayudar a descubrir conexiones sutiles; esta técnica requiere video o una secuencia rápida de placas (videofluoroscopio) para ver el llenado momentáneo y el rápido vaciamiento de la fístula como en nuestro segundo paciente (2,6,14).
La tomografía computarizada con cortes sagitales puede ser de utilidad diagnóstica en la atresia esofágica con fístula distal o sin ella para medir la distancia entre las bolsas superior e inferior en su posición natural, la cual es predictiva de la mortalidad en el procedimiento quirúrgico reconstructivo y de la morbilidad a largo plazo, permitiendo planear mejor las estrategias preoperatorias y postoperatorias en el caso de continuidad de la ventilación mecánica, pero se debe tener en cuenta el costo-beneficio de este estudio según el caso (3,16).
Otra manera de hacer el diagnóstico de FTE, sobre todo en los casos en que no se la observe en las imágenes, es con la fibrobroncoscopia con sedación manteniendo la respiración espontánea, la que permite visualizar la localización exacta de la FTE con respecto a las cuerdas vocales y la carina, determinando así el calibre y la longitud. Es de utilidad para planear un enfoque quirúrgico apropiado, así como para detectar otras alteraciones como traqueomalacia. El broncoscopio rígido se puede utilizar en estos pacientes ya que permite visualizar la fístula, ocluirla y controlar mejor la ventilación en caso de deterioro respiratorio. La canalización de la fístula se puede hacer por cualquiera de dos técnicas: por broncoscopia rígida con un catéter de Fogarty, con una guía recubierta de teflón, o con fibrobroncoscopio de 2,2 mm a través del tubo orotraqueal durante la cirugía, que por transiluminación ayuda a identificar la fístula intraoperatoriamente (6,17-19). Para mejorar el rendimiento de estas técnicas se ha utilizado el azul de metileno inyectado a través de la tráquea para identificarlo en el esófago, como se hizo en el segundo caso reportado (1,6,10). La endoscopia de vías digestivas superiores identifica la FTE con menor frecuencia debido a que los pliegues del esófago dificultan su visualización (11).
Todos los pacientes con AE con fístula o sin ella deben ser evaluados para descartar la presencia de otras malformaciones congénitas; para ello se hacen: ecocardiografía con el fin de definir la presencia de anormalidades cardíacas y de arco aórtico derecho, caso en el cual algunos cirujanos optan por un abordaje izquierdo. En la radiografía de tórax se pueden evaluar anormalidades vertebrales y pulmonares y se debe complementar el estudio con la ecografía renal y de las vías urinarias (1-3).
Entre los diagnósticos diferenciales de esta entidad se encuentran el reflujo gastroesofágico, los trastornos alimentarios o de la deglución, la hendidura laríngea, las compresiones esofágicas extrínsecas y la estenosis esofágica (1,2,14).
El cierre de la FTE depende de su ubicación: si se encuentra por encima de T2, el abordaje debe ser por cervicotomía derecha porque presenta menos riesgo de lesión del nervio laríngeo recurrente; la toracotomía se reserva para los casos en que la fístula está a nivel de T2 o por debajo, lo cual ocurre hasta en 30% de los casos; también se han reportado casos exitosos de cierre quirúrgico mediante toracoscopia (2,3,20,21). Los niños con bajo peso y otras complicaciones asociadas deben tener soporte parenteral y gastrostomía y en ellos se debe diferir la intervención quirúrgica hasta corregir estas situaciones; así se procedió en nuestro segundo paciente (2,22).
Se han desarrollado otras técnicas de cierre sobre todo en los casos de recanalización de la fístula ya que el procedimiento abierto implica mayor morbimortalidad; el cierre se lleva a cabo mediante broncoscopia: se ubica el orificio de la fístula, se hace un despulimiento de sus paredes laterales y luego se aplica fibrina, como en nuestro segundo caso, aunque sin éxito. Además, se ha reportado el uso de electrocauterización (23-27).
Las complicaciones de los pacientes con FTE luego de la corrección pueden ser agudas o crónicas: entre las primeras la más importante es la dehiscencia de las suturas que se presenta hasta en 17% de los casos; en el segundo grupo está la estenosis esofágica hasta en 50% de los pacientes, acompañada de retardo en el crecimiento (2,28).
El trastorno de la deglución se presenta en los primeros años de vida hasta en 75% a 100% de los casos, por alteraciones en el peristaltismo del esófago; se observa un segmento inmóvil o con contractilidad incoordinada, que se puede evaluar en la manometría. La enfermedad por reflujo gastroesofágico y el trastorno de la deglución con aspiración causan falla en el crecimiento hasta en 65% de los casos, principalmente en los pacientes con FTE asociada a AE (2,28). La enfermedad por reflujo gastroesofágico se presenta en 35% a 58% de los casos. Ocurre por disfunción motora intrínseca o acortamiento de la porción intraabdominal del esófago debido a la tensión de la anastomosis. Puede llevar a estenosis esofágica, aspiración y desarrollo subsecuente de neumonías y cuadros bronco-obstructivos recurrentes o persistentes. Además puede producir apneas, crisis de cianosis y falla en el crecimiento. El 56% de los pacientes responden al tratamiento médico y el resto requieren cirugía antirreflujo (28).
Las estenosis esofágicas postoperatorias se presentan con menor frecuencia en los pacientes con FTE sola. Generalmente ocurren en el esófago inferior por compromiso vascular, dada su irrigación segmentaria por la aorta y las arterias intercostales. Se manifiestan por disfagia, vómitos, baja ganancia de peso, obstrucción esofágica, aspiración del contenido deglutido y neumonías recurrentes. El tratamiento consiste en dilataciones esofágicas periódicas (1,2,25).
Las complicaciones pulmonares ocurren en 46% de los casos, generalmente son consecuencia de la enfermedad por reflujo gastroesofágico, traqueomalacia, persistencia de la FTE y estenosis esofágicas (20). Además se han reportado casos de cáncer escamocelular años después de la corrección quirúrgica, probablemente asociados a la inflamación crónica en zonas de refistulización (22,23).
La recurrencia de la FTE ocurre en 9% de los casos entre los 2 y 18 meses después de la corrección; se manifiesta por tos recurrente, ahogo, cianosis con la alimentación o neumonía recurrente. Esto es más frecuente después de la ligadura de la fístula que luego de su sección completa; otras situaciones que pueden favorecer la recurrencia son: la dehiscencia de las suturas que se presenta en 21% de los casos o las líneas de sutura traqueales y esofágicas contiguas. En el caso de la dehiscencia de suturas últimamente se ha probado con un agente sintético anticolinérgico para disminuir la secreciones y permitir que se cierren espontáneamente (28-30).
El pronóstico de esta enfermedad se basa en sus comorbilidades como cardiopatías cianógenas o bajo peso al nacer y las complicaciones respiratorias por requerimiento de ventilación mecánica.
CONCLUSIÓN
El diagnóstico de las FTE no es fácil e implica un alto índice de sospecha en neonatos con dificultad respiratoria, tos y cianosis relacionadas con los períodos de alimentación y que en ocasiones pueden llegar a la falla ventilatoria; en niños más grandes puede manifestarse con neumonía recurrente y síntomas respiratorios con la alimentación. Por esto son indispensables, para hacer el diagnóstico y tratamiento oportunos, una historia clínica detallada y estudios imaginológicos bien seleccionados e interpretados.
Conflicto de intereses y financiación
Hacemos constar que durante la realización del trabajo no se presentaron conflictos de interés, ni se requirió financiación; los cuatro autores estamos de acuerdo con el contenido del texto y este no ha sido publicado anteriormente.
REFERENCIAS BIBLIOGRÁFICAS
1. Genty E, Attal P, Nicollas R, Roger G, Triglia JM, Garabedian EN, et al. Congenital tracheoesophageal fistula without esophageal atresia. Int J Pediatr Otorhinolaryngol. 1999 May 25;48(3):231–8. [ Links ]
2. Holland AJA, Fitzgerald DA. Oesophageal atresia and tracheo-oesophageal fistula: current management strategies and complications. Paediatr Respir Rev. 2010 Jun;11(2):100–6; quiz 106–7. [ Links ]
3. Naik-Mathuria B, Olutoye OO. Foregut abnormalities. Surg Clin North Am. 2006 Apr;86(2):261–84, viii. [ Links ]
4. Shaw-Smith C. Oesophageal atresia, tracheo-oesophageal fistula, and the VACTERL association: review of genetics and epidemiology. J Med Genet. 2006 Jul;43(7):545–54. [ Links ]
5. de Jong EM, Felix JF, de Klein A, Tibboel D. Etiology of esophageal atresia and tracheoesophageal fistula: ''mind the gap''. Curr Gastroenterol Rep. 2010 Jun;12(3):215–22. [ Links ]
6. Dave S, Bajpai M, Gupta DK, Agarwala S, Bhatnagar V, Mitra DK. Esophageal atresia and tracheo-esophageal fistula: a review. Indian J Pediatr. 1999;66(5):759–72. [ Links ]
7. El-Gohary Y, Gittes GK, Tovar JA. Congenital anomalies of the esophagus. Semin Pediatr Surg. 2010 Aug;19(3):186–93. [ Links ]
8. Kinottenbelt G, Skinner A, Seefelder C. Tracheooesophageal fistula (TOF) and oesophageal atresia (OA). Best Pract Res Clin Anaesthesiol. 2010 Sep;24(3):387–401. [ Links ]
9. Felix JF, Tibboel D, de Klein A. Chromosomal anomalies in the aetiology of oesophageal atresia and tracheo-oesophageal fistula. Eur J Med Genet. 2007;50(3):163–75. [ Links ]
10. Baisi A, Bonavina L, Narne S, Peracchia A. Benign tracheoesophageal fistula: results of surgical therapy. Dis Esophagus. 1999 Jan;12(3):209–11. [ Links ]
11. Karnak I, Senocak ME, Hiçsönmez A, Büyükpamukçu N. The diagnosis and treatment of H-type tracheoesophageal fistula. J Pediatr Surg. 1997 Dec;32(12):1670–4. [ Links ]
12. Beasley SW, Myers NA. The diagnosis of congenital tracheoesophageal fistula. J Pediatr Surg. 1988 May;23(5):415–7. [ Links ]
13. Gardella C, Tomà P, Sacco O, Girosi D, Panigada S, Battistini E, et al. Intermittent gaseous bowel distention: atypical sign of congenital tracheoesophageal fistula. Pediatr Pulmonol. 2009 Mar;44(3):244–8. [ Links ]
14. Fordham LA. Imaging of the esophagus in children. Radiol Clin North Am. 2005 Mar;43(2):283–302. [ Links ]
15. Houben CH, Curry JI. Current status of prenatal diagnosis, operative management and outcome of esophageal atresia/tracheo-esophageal fistula. Prenat Diagn. 2008 Jul;28(7):667–75. [ Links ]
16. Nagata K, Kamio Y, Ichikawa T, Kadokura M, Kitami A, Endo S, et al. Congenital tracheoesophageal fistula successfully diagnosed by CT esophagography. World J Gastroenterol. 2006 Mar 7;12(9):1476–8. [ Links ]
17. Atzori P, Iacobelli BD, Bottero S, Spirydakis J, Laviani R, Trucchi A, et al. Preoperative tracheobronchoscopy in newborns with esophageal atresia: does it matter? J Pediatr Surg. 2006 Jun;41(6):1054–7. [ Links ]
18. Garcia NM, Thompson JW, Shaul DB. Definitive localization of isolated tracheoesophageal fistula using bronchoscopy and esophagoscopy for guide wire placement. J Pediatr Surg. 1998 Nov;33(11):1645–7. [ Links ]
19. Goyal A, Potter F, Losty PD. Transillumination of H-type tracheoesophageal fistula using flexible miniature bronchoscopy: an innovative technique for operative localization. J Pediatr Surg. 2005 Jun;40(6):e33–4. [ Links ]
20. Rothenberg SS. Thoracoscopic repair of tracheoesophageal fistula in newborns. J Pediatr Surg. 2002 Jun;37(6):869–72. [ Links ]
21. Aziz GA, Schier F. Thoracoscopic ligation of a tracheoesophageal H-type fistula in a newborn. J Pediatr Surg. 2005 Jun;40(6):e35–6. [ Links ]
22. Healey PJ, Sawin RS, Hall DG, Schaller RT, Tapper D. Delayed primary repair of esophageal atresia with tracheoesophageal fistula: is it worth the wait? Arch Surg. 1998 May;133(5):552–6. [ Links ]
23. Allal H, Montes-Tapia F, Andina G, Bigorre M, Lopez M, Galifer RB. Thoracoscopic repair of Htype tracheoesophageal fistula in the newborn: a technical case report. J Pediatr Surg. 2004 Oct;39(10):1568–70. [ Links ]
24. Bruch SW, Hirschl RB, Coran AG. The diagnosis and management of recurrent tracheoesophageal fistulas. J Pediatr Surg. 2010 Feb;45(2):337–40. [ Links ]
25. Meier JD, Sulman CG, Almond PS, Holinger LD. Endoscopic management of recurrent congenital tracheoesophageal fistula: a review of techniques and results. Int J Pediatr Otorhinolaryngol. 2007 May;71(5):691–7. [ Links ]
26. Farra J, Zhuge Y, Neville HL, Thompson WR, Sola JE. Submucosal fibrin glue injection for closure of recurrent tracheoesophageal fistula. Pediatr Surg Int. 2010 Feb;26(2):237–40. [ Links ]
27. Tzifa KT, Maxwell EL, Chait P, James AL, Forte V, Ein SH, et al. Endoscopic treatment of congenital HType and recurrent tracheoesophageal fistula with electrocautery and histoacryl glue. Int J Pediatr Otorhinolaryngol. 2006 May;70(5):925–30. [ Links ]
28. Kovesi T, Rubin S. Long-term complications of congenital esophageal atresia and/or tracheoesophageal fistula. Chest. 2004 Sep;126(3):915–25. [ Links ]
29. Esquibies AE, Zambrano E, Ziai J, Kesebir D, Touloukian RJ, Egan ME, et al. Pulmonary squamous cell carcinoma associated with repaired congenital tracheoesophageal fistula and esophageal atresia. Pediatr Pulmonol. 2010 Feb;45(2):202–4. [ Links ]
30. Mathur S, Vasudevan SA, Patterson DM, Hassan SF, Kim ES. Novel use of glycopyrrolate (Robinul) in the treatment of anastomotic leak after repair of esophageal atresia and tracheoesophageal fistula. Journal of pediatric surgery. 2011 Mar;46(3):e29–32. [ Links ]
