










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.27 no.3 Medellín July/Sept. 2014
PRESENTACIÓN DE CASOS
Encefalopatía espongiforme transmisible humana: reporte de un caso
Human transmissible spongiform encephalopathy: Case report
Camilo Duque-Velásquez1; Ánderson Garzón Alzate3; Andrés Villegas Lanau2; Laura Marcela Escobar Velásquez1; Julián Zea Lopera2; Francisco Lopera2; Juan David Rodas González1*
1 Grupo de Investigación en Ciencias Veterinarias (CENTAURO). Facultad de Ciencias Agrarias, Universidad de Antioquia, Medellín, Colombia.
2 Grupo de Neurociencias de Antioquia. Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
3 Colegio Mayor de Antioquia, Medellín, Colombia. juandavid.rodas@gmail.com
Sede de Investigación Universitaria (SIU), carrera 53 # 61-30, Laboratorio 233, Grupo de Investigación en Ciencias Veterinarias, Centauro, Telefax: 2196525 Universidad de Antioquia, Medellín, Colombia, AA 1226
Recibido: junio 05 de 2013
Aceptado: octubre 28 de 2013
RESUMEN
Se presenta el caso de una mujer de 64 años con un cuadro clínico de ocho meses de duración, consistente en deterioro motor y cognitivo, que progresó rápidamente. Recibió tratamiento con quinacrina sin obtener beneficios y falleció en estado terminal, por choque séptico secundario a bronconeumonía por broncoaspiración. El cerebro fue donado para investigación y su estudio histopatológico reveló la presencia de lesiones espongiformes, astrogliosis y depósitos de proteína priónica (PrPRes) confirmados por Western blot. Todos estos rasgos se consideran característicos de la enfermedad por priones. Con este caso, no solo se informa sobre una enfermedad infrecuente en la casuística colombiana, sino que por primera vez en el país se usan simultáneamente la inmunohistoquímica y el Western blot como herramientas para el diagnóstico de estas enfermedades.
PALABRAS CLAVE
Enfermedad de Creutzfeldt-Jakob, Enfermedad por Priones, Espongiosis, Inmuno-Histoquímica, Western blot
SUMMARY
We report the case of a 64 year-old woman with motor and cognitive deterioration that progressed rapidly during eight months. She was unsuccessfully treated with quinacrine, and died in a terminal status, by septic shock secondary to bronchopneumonia by broncho-aspiration. The brain was donated for research and the histopathological analysis showed spongiform changes, astrogliosis and prion protein (PrPRes) deposits, confirmed by Western blot (WB). These features are considered characteristic of prion diseases, which are uncommon in Colombia. We highlight that its diagnosis was made for the first time in this country by the simultaneous use of immunohistochemistry and Western blot.
KEY WORDS
Creutzfeldt-Jakob Disease, Immunohistochemistry, Prion Diseases, Spongiosis, Western blot
INTRODUCCIÓN
Las encefalopatías espongiformes transmisibles (enfermedades priónicas) son un grupo de trastornos neurodegenerativos fatales de los mamíferos. En humanos se han reportado varias de ellas, a saber: la enfermedad de Creuzfeldt-Jakob, presentación familiar (CJDf), esporádica (CJDe) o infecciosa (CJD). Entre las formas infecciosas se encuentran la variante (vCJD) asociada al consumo de carne de bovinos con encefalopatía espongiforme bovina vulgarmente conocida como ''enfermedad de las vacas locas'', el Kurú (enfermedad asociada a rituales caníbales de la tribu Fore en Papua Nueva Guinea) y presentaciones iatrogénicas. Otras enfermedades priónicas que afectan a los humanos incluyen: la enfermedad de Gerstmann- Sträussler-Scheincker (GSS), el insomnio fatal familiar (IFF) y el insomnio fatal esporádico (IFe) (1).
Los priones son agregados proteicos (algunos de ellos transmisibles) asociados a estas enfermedades; están compuestos de una isoforma anormalmente plegada de la proteína priónica celular (PrPC), una glicoproteína de membrana expresada en los tejidos neuronal y linfoide (2). De acuerdo con los trabajos de Stanley Prusiner (Premio Nobel de Medicina, 1997), la isoforma patológica denominada PrPRes (debido a su resistencia parcial a la digestión con proteinasa K (PK) tiene la propiedad de inducir el cambio conformacional de su isoforma celular (PrPC) con lo que altera sus propiedades fisicoquímicas, le produce resistencia parcial a la PK y la agrega a la estructura proteica en crecimiento (3).
Las enfermedades priónicas se caracterizan por presentar en el encéfalo de los individuos afectados vacuolizaciones con apariencia microscópica de esponja, astrogliosis y depósitos proteicos de PrPRes (4). Estas enfermedades se pueden adquirir de diferentes formas: 1) por infección con el agente causal (vía oral o iatrogénica); 2) por predisposición genética (mutaciones en el gen de la proteína priónica celular en células germinales); 3) de forma idiopática (por conversión espontánea de la PrPC en PrPRes o por mutación espontánea en el locus PrnP en las neuronas) (5). El 85% de los casos corresponden al CJDe cuya incidencia anual es de un caso por millón de personas. En Europa, entre 10% y 15% de los casos son de origen familiar (genético) (6,7). Para otras presentaciones menos comunes como la de GSS se calcula una prevalencia de 1-10 por cada 100.000.000 habitantes al año (8).
De acuerdo con la presentación clínica, se puede clasificar la CJD en diferentes tipos: clásica, Heidenhain, cerebelosa, panencefálica, amiotrófica o atípica. De estas presentaciones, la más común es la clásica, que constituye el 60% de los casos (9).
DESCRIPCIÓN DEL CASO
Mujer natural de Medellín y residente en Estados Unidos, ama de casa, con el antecedente de depresión desde los 49 años, sin historia familiar de enfermedades neurológicas. A los 63 años inició un cuadro clínico de trastorno de la marcha, consistente en temblor y ataxia; pocos días después le comenzaron trastornos cognitivos y alteración del lenguaje que progresó rápidamente hasta el mutismo. Se inició el estudio del caso en los Estados Unidos, donde le hicieron la primera resonancia magnética nuclear (RMN), en la que reportaron señales hiperintensas que involucraban la sustancia blanca subcortical de ambos hemisferios (cambios inespecíficos). Un mes más tarde se detectaron mioclonias generalizadas, anomia, dolor en las extremidades inferiores, disminución de la agudeza visual, marcha inestable y mareo con vértigo. Se le hizo otra RMN, que se afectó por los movimientos de la paciente durante el examen; el informe fue de imágenes cerebrales normales para la edad. El examen del líquido cefalorraquídeo fue informado como normal; también se hicieron tomografías computadas de tórax, abdomen y pelvis, y ecografía tiroidea en las que no se hallaron lesiones sugestivas de malignidad. Un mes más tarde se le hizo PET/ CT Scan con los siguientes hallazgos: moderado hipometabolismo en los lóbulos frontotemporales, que sugería demencia. En Estados Unidos se le hicieron electroencefalograma, electrocardiograma, gammagrafía ósea, hemograma, ionograma, tiempo de coagulación, BUN, creatinina, glicemia, nivel sérico de vitamina B12, perfil paraneoplásico (Hu, MA2, Yo, Ri, CV2, ZIC4 y antígeno carcinoembrionario), perfil tiroideo y radiografía de tórax, todos los cuales fueron informados como normales. Se inició tratamiento con quinacrina y se le hicieron mensualmente monitoreo de las funciones hepática y hematológica por las posibles discrasias generadas por este medicamento; se le recomendó retornar a Colombia para estar en compañía de su familia. Cinco meses después de haberse iniciado los síntomas, la paciente era dependiente en un 100% para las actividades de la vida diaria, con deterioro motor y cognitivo grave, descontrol de esfínteres y dificultad para la deglución; requería caminador o silla de ruedas para desplazarse, tenía trastornos del sueño que se iniciaron con el síndrome de piernas inquietas y presentaba alteraciones visuales y auditivas; en estas condiciones regresó a Colombia. Fue evaluada por el Grupo de Neurociencias de Antioquia y se la encontró en mutismo, con la mirada perdida y dificultad para fijarla; no respondía a las órdenes; tenía rigidez generalizada, más marcada en el hemicuerpo izquierdo, mioclonias generalizadas, hiperreflexia osteotendinosa; estaba con sonda vesical y recibía tratamiento con quinacrina 100 miligramos cada 8 horas y ácido valproico. Se le hizo una nueva RMN con y sin contraste en la que informaron: hiperintensidades de difusión restringida en los ganglios basales bilaterales, difusión restringida del cíngulo izquierdo y de la corteza frontal izquierda, sugestivos de la enfermedad de Creutzfeldt-Jakob. Un mes más tarde se tornó cianótica, diaforética y con baja saturación de oxígeno y falleció poco después con el diagnóstico de choque séptico secundario a bronconeumonía por broncoaspiración.
Al fallecer la paciente, sus familiares, previa firma del consentimiento informado, donaron su encéfalo, con fines de investigación, al Neurobanco del Grupo de Neurociencias de Antioquia. Se hizo viscerotomía y se extrajo el encéfalo siguiendo las normas de bioseguridad, de acuerdo con la reglamentación establecida para estos fines por la OMS (10). Una vez obtenido, el encéfalo fue inactivado en ácido fórmico al 95% durante una hora y luego fijado en formaldehído bufferizado al 10%, en el que permaneció durante 20 días para luego obtener muestras para estudio histopatológico.
Los hallazgos del estudio macroscópico fueron los siguientes: peso del encéfalo 953,3 gramos; peso del contenido de la fosa posterior 101,4 gramos. Líquido cefalorraquídeo, meninges y sistema vascular de aspecto normal; cerebro congestivo, con atrofia global grave, leve borramiento de la corteza cerebral en los lóbulos temporales, punteado fino de color blanco en los núcleos caudado y lenticular y tálamo reducido de tamaño. El cerebelo también tenía el tamaño reducido especialmente a expensas de las folias; el tallo cerebral estaba moderadamente reducido de tamaño y la pigmentación de la sustancia nigra era normal.
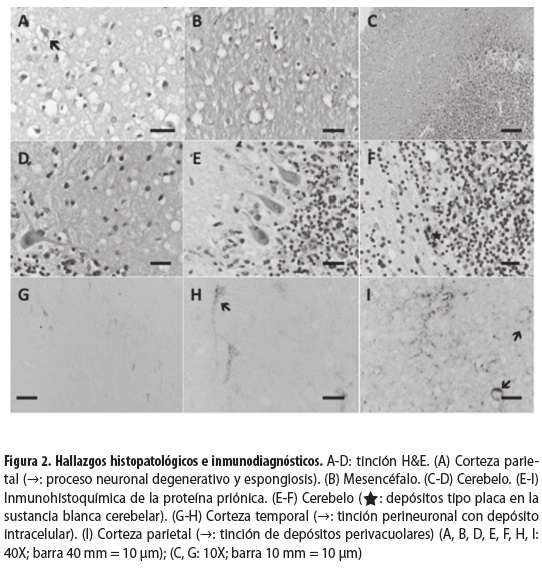
En el estudio histológico del tejido se demostró gliosis por medio de la tinción de la proteína ácida glial fibrilar (GFAP) (figura 1); con la tinción de hematoxilina- eosina (HE) se observaron lesiones compatibles con espongiosis, en forma de vacuolas en la corteza del lobulillo parietal superior; estas lesiones también se observaron en la corteza occipital. En ambos casos se pudieron observar procesos neuronales degenerados (figura 2A); también se encontraron: degeneración espongiforme en el mesencéfalo (figura 2B), pérdida celular en la capa granular del cerebelo, alteración de la sustancia blanca y de la capa de células de Purkinje y múltiples vacuolas en la capa molecular (figuras 2C y 2D). El análisis inmunohistoquímico del tejido reveló la presencia de depósitos de la proteína priónica con diferentes patrones de tinción: difusos, granulares y focalizados (tipo placa) (figuras 2E y 2I). Se observaron además depósitos perivacuolares, perineuronales e intracitoplasmáticos.
El estudio del tejido encefálico por Western blot reveló la presencia de fragmentos resistentes a la degradación por la proteinasa K; dichos fragmentos representan tres diferentes glicoformas (PrP di-, mono- y sin glicosilar) de menor peso molecular debido a la pérdida de la región N-terminal de la proteína por la digestión enzimática (figura 3).
DISCUSIÓN
De acuerdo con el cuadro clínico de esta paciente, se deben tener en cuenta varios diagnósticos diferenciales tales como: un síndrome paraneoplásico, infecciones virales o bacterianas, contacto con agentes tóxicos u otros trastornos neurodegenerativos como enfermedad de Alzheimer, demencia por cuerpos de Lewy o demencia frontotemporal. Sin embargo, factores como el tiempo de evolución, los resultados de los exámenes de laboratorio y los del estudio histopatológico permitieron excluir tales enfermedades (11).
Según los criterios estándar establecidos internacionalmente para el diagnóstico de la CJDe, podemos afirmar que nuestra paciente cumplía las condiciones de un posible caso de CJDe (12,13) y que presentó la forma clásica de esta; aunque algunos de los síntomas observados son característicos de las formas cerebelosa y de Heindenhain, los síntomas visuales y la preponderancia de los síntomas cognitivos sobre los motores no fueron lo suficientemente intensos como para clasificarla en una de estas variantes (14-16).
Nuestros hallazgos permiten confirmar que esta paciente representa un caso de enfermedad por priones, pero no es posible por el momento determinar la forma en que la adquirió, puesto que no tenía historia familiar de CJD, lo cual podría indicar que no se trata de un caso de origen genético. Sería recomendable hacer un estudio del gen de la proteína priónica en busca de mutaciones que hayan podido predisponer a la conversión de la isoforma celular en la isoforma anormal y parcialmente resistente a la proteinasa K, puesto que se ha descrito un número importante de mutaciones en el gen Prnp en individuos que han sufrido la enfermedad sin tener historia familiar de ella. También es recomendable hacer un estudio genético en pacientes con una sintomatología similar, para determinar la presencia del polimorfismo en el codón 129 y establecer el genotipo para metionina o valina, que ha sido propuesto como factor de susceptibilidad con repercusiones en la sintomatología de los pacientes (17,18).
La paciente vivió durante un tiempo prolongado por fuera del país, lapso durante el cual pudo haberse expuesto al contacto con el agente causante de la encefalopatía espongiforme bovina, pero no cumplía los criterios diagnósticos para clasificarla como un caso de la variante de CJD (vCJD) (13,19). De acuerdo con los cambios observados en el tejido encefálico mediante la tinción con hematoxilina-eosina, estos hallazgos son más compatibles con los reportados para casos esporádicos, iatrogénicos y genéticos ''familiares'' de CJD (20). La tinción inmunohistoquímica para la proteína priónica en el tejido encefálico reveló depósitos compatibles con lo informado en la literatura para la neuropatología de la CJD (21). La presencia de depósitos de tipo placa se ha restringido en el pasado a casos de etiología iatrogénica, familiar y de la variante de Creutzfeldt-Jakob, pero existen informes de este tipo de depósitos en casos de etiología esporádica (22). La presencia de fragmentos de PrPRes resistentes a la digestión por PK constituye la prueba confirmatoria de un caso de demencia por priones; de esta forma se cumple con los criterios internacionales establecidos para el diagnóstico definitivo de CJDe (23).
En Colombia existe el reporte de un caso de scrapie (la enfermedad clásica por priones que afecta al ganado ovino) y en humanos se han informado varios casos de CJD (24). Hasta la fecha no se han reportado casos de encefalopatía espongiforme bovina, ni de la variante CJD (vCJD). Nuestro caso, además de ayudar al conocimiento de esta enfermedad en Colombia, es el primero que empleó todas las técnicas estándar que se hacen en el país. Además del estudio inmunohistoquímico para las lesiones típicas compatibles con la enfermedad, hemos suministrado evidencia por Western blot del mismo caso, lo que abre las puertas al estudio de otros que se presenten en el país y a su caracterización molecular. El estudio de dichos mecanismos es de gran importancia no solo para la investigación local sobre el tema, sino también para el estudio epidemiológico de los casos; además, los resultados obtenidos podrían también ser pertinentes para la investigación internacional.
En conclusión, se debe considerar el diagnóstico de CJD en todo paciente que presente un trastorno cognitivo, sea leve o grave, con deterioro rápido, acompañado de alteraciones motoras y al que se le descarten otras causas posibles de sus síntomas (25).
AGRADECIMIENTOS
Expresamos nuestra gratitud a Colciencias por el apoyo económico a este estudio mediante el proyecto ''Estandarización de técnicas para el diagnóstico de enfermedades priónicas en muestras de tejido nervioso humano'' Código: 111540820493. Además, agradecemos al doctor Christopher Johnson (USGS-National Wildlife Health Center, Madison, Wisconsin, USA) por la gentil donación de anticuerpos y controles positivos para inmunohistoquímica.
CONFLICTOS DE INTERÉS
Los autores declaran que no existe ningún conflicto de interés con los contenidos enunciados en este artículo.
RESPONSABILIDADES DE LOS AUTORES
El estudio se llevó a cabo en la Sede de Investigaciones Universitarias de la Universidad de Antioquia. Los doctores Francisco Lopera y Andrés Villegas tuvieron a su cargo la obtención de la información clínica y la evaluación de la paciente; los investigadores Julián Zea, Camilo Duque-Velásquez y Andrés Villegas obtuvieron las muestras; los investigadores Camilo Duque- Velásquez, Ánderson Garzón, Andrés Villegas, Laura Escobar y Juan Rodas procesaron las muestras. Los investigadores Juan Rodas y Andrés Villegas dirigieron el estudio del caso y la totalidad de los autores intervinieron en la elaboración del manuscrito.
REFERENCIAS BIBLIOGRÁFICAS
1. Dalsgaard NJ. Prion diseases. An overview. APMIS. 2002;110(1):3–13. [ Links ]
2. Chesebro B. Introduction to the transmissible spongiform encephalopathies or prion diseases. Br Med Bull. 2003;66:1–20. [ Links ]
3. Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363–83. [ Links ]
4. Budka H. Neuropathology of prion diseases. Br Med Bull. 2003;66:121–30. [ Links ]
5. Prusiner SB. Prion diseases and the BSE crisis. Science. 1997;278(5336):245–51. [ Links ]
6. Pedersen NS, Smith E. Prion diseases: epidemiology in man. APMIS. 2002; 110(1):14-22. [ Links ]
7. Roeber S, Krebs B, Neumann M, Windl O, Zerr I, Grasbon- Frodl E-M, et al. Creutzfeldt-Jakob disease in a patient with an R208H mutation of the prion protein gene (PRNP) and a 17-kDa prion protein fragment. Acta Neuropathol. 2005;109(4):443–8. [ Links ]
8. Liberski PP. Spongiform change--an electron microscopic view. Folia Neuropathol. 2004; 42 (Suppl B):59-70. [ Links ]
9. Zivkovic S, Boada M, Lopez O. [Review of Creutzfeldt- Jakob disease and other prion diseases]. Rev Neurol. 2000; 31(12):1171-1179. [ Links ]
10. World Health Organization. WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation, Geneva, Switzerland, 23-26 March 1999. Geneva: WHO; 1999. [ Links ]
11. Paterson RW, Torres-Chae CC, Kuo AL, Ando T, Nguyen EA, Wong K, et al. Differential diangosis of Jakob- Creutzfeldt Disease. Arch Neurol. 2012; 69(12):1578- 1582. [ Links ]
12. Weber T, Otto M, Bodemer M, Zerr I. Diagnosis of Creutzfeldt-Jakob disease and related human spongiform encephalopathies. Biomed Pharmacother. 1997; 51(9):381-387. [ Links ]
13. World Health Organiztion. Department of communicable disease surveillance and response. 2nd ed. Geneve: World Health Organiztion; 1999. [ Links ] 14. Armstrong RA. Creutzfeldt-Jakob disease and vision. Clin Exp Optom. 2006;89(1):3–9. [ Links ]
15. Rabinovici GD, Wang PN, Levin J, Cook L, Pravdin M, Davis J, et al. First symptom in sporadic Creutzfeldt- Jakob disease. Neurology. 2006;66(2):286–7. [ Links ]
16. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132(Pt 10):2659–68. [ Links ]
17. Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14(3):273–81. [ Links ]
18. Villegas A. Enfermedades por priones: de la clínica a la biología molecular. Acta Neurol Colomb. 2010; 26(2):87-111. [ Links ]
19. Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol. 2000;47(5):575–82. [ Links ]
20. Iwasaki Y, Mimuro M, Yoshida M, Hashizume Y, Kitamoto T, Sobue G. Clinicopathologic characteristics of five autopsied cases of dura mater-associated Creutzfeldt-Jakob disease. Neuropathology. 2008;28(1):51–61. [ Links ]
21. Bell JE, Gentleman SM, Ironside JW, McCardle L, Lantos PL, Doey L, et al. Prion protein immunocytochemistry-- UK five centre consensus report. Neuropathol Appl Neurobiol. 1997;23(1):26–35. [ Links ]
22. Tanaka S, Saito M, Morimatsu M, Ohama E. Immunohistochemical studies of the PrP(CJD) deposition in Creutzfeldt-Jakob disease. Neuropathology. 2000;20(2):124–33. [ Links ]
23. Hill AF, Joiner S, Beck JA, Campbell TA, Dickinson A, Poulter M, et al. Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain. 2006;129(Pt 3):676–85. [ Links ]
24. Duque-Velásquez JC, Villegas A, Rodas JD. Encefalopatías espongiformes transmisibles: biología del prion y estado actual de la vigilancia epidemiológica en Colombia. Rev Colomb Cienc Pecu. 2010; 23: 240-249. [ Links ]
25. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: Dsm- 5. Arlington: Amer Psychiatric Pub Incorporated; 2013. [ Links ]
