










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.27 no.4 Medellín Oct./Dec. 2014
ARTÍCULO DE REVISIÓN
Pruebas bioquímicas para la detección de metabolitos producidos en los errores innatos del metabolismo
Biochemical tests for the detection of metabolites produced in inborn errors of metabolism
Provas bioquímicas para a detecção de metabólitos produzidos nos erros inatos do metabolismo
Natalia Regina Mesa Herrera1; Cristian Andrés Carmona Carmona2; Luis Carlos Burgos Herrera3
1 Profesora de Bioquímica y de Errores Innatos del Metabolismo, Departamento de Fisiología y Bioquímica, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. natalimesa@yahoo.com
2 Estudiante de Microbiología y Bioanálisis, Escuela de Microbiología, Universidad de Antioquia, Medellín, Colombia.
3 Profesor de Bioquímica y de Errores Innatos del Metabolismo, Departamento de Fisiología y Bioquímica, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
Recibido: noviembre 18 de 2013
Aceptado: febrero 20 de 2014
RESUMEN
Los errores innatos del metabolismo (EIM) son más de 550 enfermedades en las que se presenta una deficiencia o ausencia de proteínas con actividad enzimática, transportadora, receptora o estructural. Cada una de estas enfermedades es rara, pero su gran variedad hace que, consideradas en conjunto, sean la principal patología neonatal. Para la detección de los metabolitos producidos en los EIM se pueden utilizar pruebas cualitativas. Su utilidad radica en que son muy rápidas y de fácil acceso, y en que sirven como pruebas presuntivas para proceder a hacer exámenes más especializados o para enfocar el diagnóstico. Teniendo en cuenta su importancia para un diagnóstico temprano de los EIM, el objetivo del presente artículo es describir el funcionamiento de las pruebas bioquímicas de resorcinol, dinitrofenilhidrazina, nitrosonaftol, nitroprusiato y Hoesch, haciendo énfasis en los metabolitos que detectan.
PALABRAS CLAVE
Errores Innatos del Metabolismo, Nitroprusiato
SUMMARY
Inborn errors of metabolism (IEM) are more than 550 diseases in which there is a deficiency or absence of proteins with enzymatic, transporter, receptor or structural activity. Individually these diseases are rare, but because of their wide variety they are, considered together, the largest neonatal disease. To detect metabolites produced in IEM qualitative tests can be used. They are easily accessible and fast to carry out, and serve as presumptive elements before proceeding to more specialized tests or to focus diagnosis. Given their importance for the early diagnosis of IEM, this article aims to describe the functioning of the following biochemical tests: dinitrophenylhydrazine, resorcinol, nitrosonaphtol, nitroprusside and Hoesch, emphasizing in the metabolites that they detect.
KEY WORDS
Inborn Errors of Metabolism, Nitroprusside
RESUMO
Os erros inatos do metabolismo (EIM) são mais de 550 doenças nas que se apresenta uma deficiência ou ausência de proteínas com atividade enzimática, transportadora, receptora ou estrutural. Cada uma destas doenças é rara, mas sua grande variedade faz que, consideradas em conjunto, sejam a principal patologia neonatal. Para a detecção dos metabólitos produzidos nos EIM se podem utilizar provas qualitativas. Sua utilidade radica em que são muito rápidas e de fácil acesso, e em que servem como provas presuntivas para proceder a fazer exames mais especializados ou para enfocar o diagnóstico. Tendo em conta sua importância para um diagnóstico precoce dos EIM, o objetivo do presente artigo é descrever o funcionamento das provas bioquímicas de resorcinol, dinitrofenilhidrazina, nitrosonaftol, nitroprusiato e Hoesch, fazendo ênfases nos metabólitos que detectam.
PALAVRAS IMPORTANTES
Dinitrofenilhidrazina; Erros Inatos do Metabolismo, Hoesh; Nitroprusiato, Nitrosonaftol, Resorcinol
INTRODUCCIÓN
Los errores innatos del metabolismo (EIM) son más de 550 enfermedades en las que se presenta deficiencia o ausencia de proteínas que tienen actividad enzimática, transportadora, receptora o estructural, lo que causa una interrupción en cualquiera de las rutas metabólicas de los azúcares, lípidos, aminoácidos, hormonas, vitaminas, entre otras (1). Como consecuencia, se pueden producir tres efectos: acumulación del sustrato, carencia del producto o activación de rutas metabólicas alternativas con producción de metabolitos secundarios tóxicos (2).
De forma individual, las enfermedades metabólicas hereditarias son raras, pero su gran variedad (más de 550) hace que, consideradas en conjunto, sean la principal patología neonatal (3). En todo el mundo se presentan en 1 de cada 2.500 recién nacidos (4). Es probable, inclusive, que su incidencia y prevalencia estén subvaloradas, debido a que muchas de ellas permanecen sin diagnosticar.
El laboratorio permite analizar tres factores para el diagnóstico de los EIM: la actividad de la enzima defectuosa, los metabolitos secundarios y el sustrato que no logró metabolizarse (5). Para el análisis se utilizan pruebas cuantitativas y cualitativas. Las primeras, como la cromatografía líquida de alto rendimiento (HPLC), son muy exactas (6); sin embargo, estos métodos cuantitativos son costosos y para hacerlos se necesitan instalaciones especiales y personal entrenado.
Entre las pruebas cualitativas, las más usadas son las bioquímicas o de tubo. En ellas se usan sustancias químicas que reaccionan con moléculas específicas para producir precipitados o compuestos coloreados, que se pueden evidenciar a simple vista (7). Su utilidad radica en que son muy rápidas y de fácil acceso, y en que sirven como pruebas presuntivas para proceder a exámenes más especializados o para enfocar mejor el diagnóstico. El presente artículo tiene como objetivo describir el funcionamiento de las pruebas bioquímicas de resorcinol, dinitrofenilhidrazina, nitrosonaftol, nitroprusiato y Hoesch, haciendo énfasis en los metabolitos que detectan.
REQUERIMIENTOS DE LAS MUESTRAS BIOLÓGICAS PARA LAS PRUEBAS
Para cada una de las pruebas que se describirán es necesaria una muestra de 1 mL de orina, preferiblemente de la primera micción matinal, que se debe transportar y almacenar a una temperatura menor de 8 °C, protegiéndola de la luz. Si la orina presenta precipitados se la debe centrifugar a 500 g, por 3 minutos, y luego filtrarla (8). Los procedimientos se pueden ver en la tabla 1.
PRUEBA DE RESORCINOL PARA LA DETECCIÓN DE FRUCTOSA
La fructosa es una cetohexosa que se encuentra libre en frutos maduros, en otros órganos vegetales y en la miel. La ingesta diaria de este carbohidrato es aproximadamente de 100 g. El trastorno de su metabolismo puede ser de dos tipos: fructosuria esencial e intolerancia hereditaria a la fructosa. La primera es una enfermedad benigna y asintomática causada por la deficiencia de la enzima fructoquinasa. En contraste, la intolerancia hereditaria a la fructosa, debida a la deficiencia de la enzima fructosa 1,6 bifosfato aldolasa, se caracteriza por marcada hipoglicemia y vómito constante. En personas que presentan esta enfermedad, una ingesta prolongada de fructosa puede provocar un daño hepático grave que cursa con hemorragia, hepatomegalia, ictericia e inclusive la muerte (9).
Las personas afectadas presentan azúcares reductores en la orina, a expensas de la fructosa. La fructosuria dependerá del tiempo y de la cantidad de fructosa o sacarosa consumida (10). Se puede detectar la fructosa en la orina mediante la prueba de resorcinol, basada en la producción de derivados del furfural (aldehído aromático) (11).
Cuando la fructosa disuelta en agua es sometida a un calentamiento a 100 °C con ácidos diluidos como el clorhídrico (HCl), se deshidrata formando hidroximetilfurfural; este compuesto se combina rápidamente con el resorcinol para formar un producto de color rojo brillante (12). En la figura 1 se muestra la reacción.
Interpretación del resultado
La prueba de resorcinol da positiva con concentraciones de fructosa mayores de 30 mmol/L (13). En los individuos que no presentan fructosuria, la concentración de fructosa está por debajo de 30 mmol/L o inclusive ausente (14). En pacientes diabéticos que presentan una marcada glucosuria, la prueba puede producir un color levemente rosado, que no se debe confundir con un resultado positivo (15).
Aunque la reacción de resorcinol se utiliza principalmente para la identificación de fructosa, también puede ocurrir con otras cetohexosas e inclusive con aldohexosas (16). La característica que permite el reconocimiento diferencial de la fructosa es el tiempo de formación del hidroximetilfurfural, que es más prolongado con otras cetohexosas y con las aldohexosas.
La prueba de resorcinol se utiliza solo para identificar monosacáridos; sin embargo, cuando en la muestra hay disacáridos también se puede obtener un color rojo, pero ello requiere más tiempo. Esta situación se presenta principalmente cuando los disacáridos tienen fructosa en su estructura, debido a que la acción del ácido puede hidrolizar los enlaces glucosídicos (17). Por ejemplo, la sacarosa produce color rojo porque está compuesta por una molécula de glucosa y una de fructosa. En cambio, disacáridos como la maltosa solo alcanzan a producir un color rosado en un tiempo largo, porque el producto de su hidrólisis son aldosas; el color rosado se debe a que la glucosa en períodos mayores de 15 minutos, bajo las condiciones de la prueba, se puede isomerizar a fructosa (12).
PRUEBA DE NITROSONAFTOL PARA LA DETECCIÓN DE TIROSINA Y DE PRODUCTOS DERIVADOS DE SU METABOLISMO
La L-tirosina es un aminoácido aromático obtenido de la dieta o por la hidroxilación del aminoácido fenilalanina; por esta razón, se la considera un aminoácido semiesencial (18). Su degradación ocurre en el citoplasma de los hepatocitos, y sirve como sustrato inicial para la síntesis de diferentes moléculas en el organismo como las catecolaminas, las hormonas tiroideas y la melanina. La velocidad de degradación de la tirosina está determinada por la actividad de la enzima tirosina aminotransferasa (19).
Las enfermedades relacionadas con el catabolismo de la tirosina se denominan hipertirosinemias, producidas por una deficiencia de las enzimas que participan en su degradación. La tirosinemia oculocutánea se debe a una deficiencia de la tirosina aminotransferasa, y da lugar a síntomas relacionados con lesiones oculares (fotofobia y erosiones corneales) y cutáneas (queratosis) (18). Mutaciones en el gen que codifica para la 4-hidroxifenilpiruvato dioxigenasa producen la hawkinsinuria, que se caracteriza por acidosis metabólica y retardo del crecimiento. La tirosinemia transitoria del recién nacido también se debe a deficiencia de esta enzima (20). Por último, la deficiencia de fumarilacetoacetato hidrolasa desencadena la tirosinemia hepatorrenal, cuyos síntomas son, entre otros, daño hepático, cirrosis, síndrome de Fanconi y neuropatías (21). Es importante mencionar que la mayoría de los síntomas de estas alteraciones se reducen cuando al enfermo se le suministra una dieta restringida en tirosina y en fenilalanina (22), de ahí la importancia de un diagnóstico temprano de la enfermedad.
En las hipertirosinemias se observa aumento de la concentración urinaria del aminoácido tirosina y de algunos productos secundarios de su metabolismo como el 4-hidroxifenilacetato y el 4-hidroxifenil-lactato (18). Estos metabolitos se pueden detectar mediante la prueba de nitrosonaftol. En esta, la interacción entre el 1-nitroso-2-naftol y varios fenoles p-sustituidos, en presencia de ácido nítrico y nitrito de sodio, forma un compuesto de color naranja característico (23). Las tres moléculas de interés biológico que reaccionan con el 1-nitroso-2-naftol son los indoles sustituidos, los derivados de guaiacol (2-metoxifenol) y los fenoles p-sustituidos. Para las tres moléculas el mecanismo de la reacción es similar (figura 2) (24).
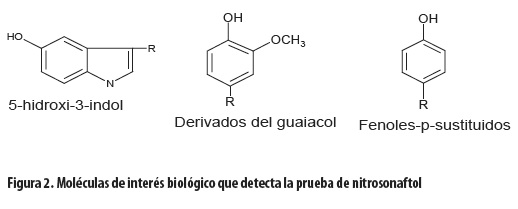
La tirosina y otros fenoles p-sustituidos reaccionan con el 1-nitroso-2-naftol produciendo un compuesto soluble de color naranja claro, cuya intensidad depende de la concentración de nitrosonaftol y de los fenoles p-sustituidos. En esta reacción el grupo químico más importante es el hidroxi. La reacción no ocurre si el grupo p-funcional es un aceptor de electrones, como, por ejemplo, el ácido 4- hidroxibenzoico (25).
Los dos reactivos necesarios, además del 1-nitroso- 2-naftol, son el ácido nítrico y el nitrito de sodio. Este último es fundamental para que se produzca la reacción, pero esta aumenta de velocidad 6 a 8 veces siempre que se agrega el ácido nítrico, ocurriendo una posible reacción de nitración en el complejo formado por el 1-nitroso-2-naftol y la sustancia que se va a identificar (26). El orden en que se añaden los reactivos afecta la velocidad de la reacción, por ejemplo, cuando se añade el ácido nítrico antes que el 1-nitroso- 2-naftol, la reacción es muy lenta y el producto final no es muy evidente, presumiblemente porque los componentes ya han sido nitrados y no permiten la reacción con el 1-nitroso-2-naftol. Según lo anterior, el ácido nítrico funciona principalmente como catalizador en la reacción y el nitrito de sodio, como un coadyuvante para la formación del complejo coloreado formado por 1-nitroso-2-naftol (24).
Interpretaciones del resultado
Un resultado positivo en la prueba de nitrosonaftol (un compuesto final de color rojo) sugiere la presencia en la orina de tirosina o sus análogos como 4-hidroxifenilpiruvato, 4-hidroxifenilacetato y 4-hidroxifenil-lactato (27). Puede haber resultados falsos positivos en los siguientes casos: tirosinemia transitoria del recién nacido, debido a que todavía no están desarrolladas las enzimas hepáticas que participan en el metabolismo de la tirosina (18); cuando están aumentadas las moléculas como el 4-hidroxifenilacetato, como ocurre en los trastornos de mala absorción o debido al metabolismo bacteriano intestinal; en personas con tumores que pueden aumentar la excreción de los ácidos homovalínico y 5-hidroxi-indolacético, productos finales del metabolismo de la dopamina y la serotonina; estas sustancias pueden producir un color rosado o púrpura en la prueba de nitrosonaftol (8).
PRUEBA DE DINITROFENILHIDRAZINA PARA LA DETECCIÓN DE ALFA-CETOÁCIDOS
En varios EIM se excretan alfa-cetoácidos en la orina. Por ejemplo, en la fenilcetonuria, en la que se presenta un defecto de la enzima fenilalanina hidoxilasa o en la producción de su cofactor la tetrahidrobiopterina. En esta enfermedad no es posible la conversión del aminoácido fenilalanina a tirosina y, en consecuencia, la fenilalanina se convierte en metabolitos secundarios como fenilpiruvato, fenilacetato y fenil-lactato, que se excretan en la orina (28). Las personas con fenilcetonuria que no tengan restricción de fenilalanina en la dieta pueden sufrir daños irreversibles del sistema nervioso central (29). Otras enfermedades relacionadas con la producción de compuestos alfa-cetoácidos en la orina son la tirosinosis, la mala absorción de la metionina (síndrome de Oasthouse), la enfermedad de la orina con olor a jarabe de arce (MSDU, por la sigla en inglés de maple syrup urine disease), la histidinemia y la tirosiluria (30).
Los compuestos alfa-cetoácidos en la orina se pueden detectar con la prueba de dinitrofenilhidrazina. Debido al grupo carbonilo, estos compuestos pueden reaccionar con la 2,4-dinitrofenilhidrazina, en medios acuosos ligeramente ácidos, dando como producto las fenilhidrazonas (figura 3) (31). Aunque al final de la reacción puedan existir grupos potencialmente activos para una posible segunda sustitución, la fenilhidrazona es lo suficientemente insoluble y se precipita en la solución antes de que se adicione una segunda molécula de fenilhidrazina (32). La reacción que permite la formación de fenilhidrazonas se da por una adición nucleofílica catalizada por el ácido, en la que ocurren una protonación del grupo carbonilo y una adición del agente nucleofílico (2,4 dinitrofenilhidrazina) al carbono, dando como producto un alcohol (33).
Las osazonas o fenilhidrazonas son compuestos sólidos con puntos de fusión definidos, que se evidencian a simple vista como un precipitado de color naranjaamarillo al final de la reacción de dinitrofenilhidrazina. Cada ozasona tiene una forma cristalina específica que depende del compuesto que se está analizando; los cristales se pueden observar al microscopio óptico (34).
Interpretaciones del resultado
Para todos los resultados positivos es necesario asegurarse de que el producto final de la reacción no se debió a un falso positivo. Para esto se dispensa 1 mL de la orina del paciente en un tubo de ensayo y se le añade 1 mL de ácido clorhídrico 2 N. Si se forma un precipitado luego de transcurridos 10 minutos, el resultado se interpreta como un falso positivo. Entre las sustancias con baja solubilidad en ácido que pueden interferir con la prueba están la mandelamina (un antibiótico) y el material de contraste radiopaco. Estas sustancias formarán un precipitado inmediatamente después de que se adicione la 2,4 dinitrofenilhidrazina (8). La presencia de acetona también da un resultado positivo, pero en este caso se puede deber a un trastorno metabólico como una hiperglicemia, una acidemia isovalérica o una enfermedad de almacenamiento de glucógeno (35).
La prueba puede resultar negativa en la orina de las personas que se encuentran en tratamiento o están afectadas parcialmente. Por el contrario, es positiva en los pacientes con niveles de fenilalanina en la sangre por encima de 1 mmol/L (indicativo de fenilcetonuria) y en aquellos con niveles de leucina en la sangre por encima de 0,8 mmol/L (35).
PRUEBA DE NITROPRUSIATO PARA LA DETECCIÓN DE CISTEÍNA
La cisteína es un aminoácido en cuya estructura química hay un grupo sulfhidrilo, que le permite interaccionar con otras moléculas, por ejemplo, una cisteína puede formar un puente disulfuro con otra cisteína, dando lugar a una cistina (36). La cisteína, junto con los aminoácidos dibásicos, es absorbida por las células epiteliales (como las de los túbulos renales) mediante un sistema de transporte luminal, para luego ser llevada a los tejidos. Los defectos en los sistemas de transporte de la membrana celular se pueden evidenciar por aminoaciduria y una concentración de aminoácidos normal o baja en el plasma (37).
Entre las aminoacidurias está la cisteinuria en la que hay mala absorción de cisteína en el intestino y reabsorción disminuida en el riñón. La excreción de cisteína en la orina depende de si el individuo es homocigoto o heterocigoto para la mutación que lleva a la deficiencia en el transporte del aminoácido (37). Por su grupo sulfhidrilo se puede identificar la cisteína en una solución acuosa mediante la prueba de nitroprusiato. Este método también permite identificar la homocisteína, precursora de la cisteína en los organismos (38).
Los reactivos necesarios para la prueba de nitroprusiato o test de Brand son el nitroprusiato de sodio y el cianuro de sodio. La reacción depende de la molécula que se pretende identificar, ya sea que esté libre o formando un compuesto. En el caso, por ejemplo, de la cistina, al añadir los reactivos de la prueba de nitroprusiato ocurre una reacción que se puede explicar en dos etapas: en la primera, durante los diez minutos iniciales, el cianuro reduce la cistina, de esta manera se rompe el enlace disulfuro y se forman dos aminoácidos de cisteína que tienen grupos sulfhidrilo libres. En la segunda etapa, el nitroprusiato de sodio interacciona con la cisteína, produciendo un compuesto soluble de color rojo-púrpura (39). Si se desea detectar solo homocisteína en una muestra de orina, se debe agregar, además, el reactivo de nitrato de plata y se produce un complejo final de color magenta (40) (figura 4).
Interpretación del resultado
Para la prueba se debe evitar la contaminación bacteriana de la orina y es necesario que esta tenga pH neutro; las muestras preservadas con ácido pueden dar resultados falsos positivos. Algunas sustancias como la N-acetilcisteína, el 2-mercaptoetanol sulfonato, el captopril, los metabolitos de la penicilina y el acetoacetato pueden producir resultados falsos positivos (38). Esta prueba es muy útil para la detección de individuos heterocigotos que sufren de cisteinuria tipos 2 y 3 (41).
PRUEBA DE HOESCH PARA LA DETECCIÓN DE PORFOBILINÓGENO
Las porfirias son un grupo de enfermedades que resultan de defectos en la biosíntesis del grupo hemo. La disminución de la actividad de las enzimas implicadas en dicha biosíntesis, por alteraciones externas o defectos genéticos, tiene como resultado una acumulación de los sustratos de las enzimas defectuosas, los cuales se excretan en la orina y las heces (42). En los individuos heterocigotos para la deficiencia enzimática, el alelo normal continúa codificando para la enzima, por lo que pueden seguir sintetizando el grupo hemo, aunque la actividad enzimática se reduzca al 50%. Según la enzima afectada, las personas pueden presentar síntomas neuroviscerales, cutáneos o ambos (43). Se debe enfatizar en que no todos los pacientes con el defecto enzimático presentan los síntomas; ello indica que existen otros factores relevantes en la expresión de la enfermedad, como la ingestión de algunos medicamentos o la demanda del grupo hemo (44). La más común de las porfirias es la cutánea tarda por deficiencia adquirida o hereditaria de la uroporfirinógeno descarboxilasa. Mientras que la porfiria aguda más frecuente es la intermitente, por deficiencia de la porfobilinógeno deaminasa (o hidroximetilbilano sintasa) (45).
En la porfiria aguda intermitente ocurren ataques agudos que afectan a los sistemas nerviosos central, periférico y autónomo, produciendo síntomas como agitación, confusión, dolor abdominal, vómitos y alteraciones sensoriales, entre otros (44). Estos ataques se asocian con agentes precipitantes como algunos medicamentos (sulfonamidas y barbitúricos), la ingestión de alcohol, el estrés o la restricción calórica importante. Durante los ataques agudos se excretan en la orina altas concentraciones de porfobilinógeno y en menor medida de ácido aminolevulínico. Sin embargo, en ausencia de ataques agudos la concentración urinaria de estos metabolitos intermediarios puede ser indetectable (46).
Para una detección rápida de porfobilinógeno en la orina, cuando el paciente está sufriendo un ataque agudo, se puede hacer la prueba de Hoesch, también llamada reacción inversa de Ehrlich. En esta prueba el anillo pirrol del porfobilinógeno se condensa en una solución ácida con el reactivo de Ehrlich (4-dimetilaminobenzaldehído), formando un compuesto de color rojo cereza, que se produce de forma instantánea luego de adicionar el reactivo a la muestra con porfibilinógeno y cuya intensidad puede disminuir con el tiempo (47). Tal disminución se debe a que el compuesto final puede presentar una segunda reacción con otra molécula de porfibilinógeno, formando un compuesto incoloro. Para evitar que la segunda reacción se produzca muy rápido, se debe mantener la condición ácida del medio en el cual se da la reacción; igualmente, el reactivo de Ehrlich debe estar en exceso mientras que el volumen de la muestra de orina debe ser menor (48). De esta manera, se espera que el 4-dimetilaminobezaldehído reaccione con todas las moléculas presentes de porfobilinógeno (figura 5).
Interpretaciones del resultado
La prueba de Hoesch es positiva a concentraciones de porfobilinógeno por encima de 10 mg/L, como ocurre en los pacientes con porfiria intermitente aguda. La excreción fisiológica de porfobilinógeno es menor de 10 mg/L y, en consecuencia, no se esperan resultados positivos en personas que no estén en un episodio agudo de porfiria o no sufran la enfermedad (49). El urobilinógeno a una concentración de hasta 200 mg/L no da un resultado positivo (50). Los indoles en la orina pueden producir un compuesto coloreado similar al porfobilinógeno con el reactivo de Ehrlich; estos compuestos se pueden deber a enfermedades relacionadas con el metabolismo de la tirosina; por ello, si los síntomas lo sugieren, se puede hacer un análisis de las muestras con la prueba de nitrosonaftol (8).
CONCLUSIONES
Las pruebas cualitativas constituyen una valiosa ayuda para el diagnóstico de muchos EIM, debido a su sencillez, facilidad de ejecución y celeridad en el reporte de los resultados. Estas pruebas permiten orientar el diagnóstico presuntivo de estas enfermedades hacia exámenes confirmatorios más especializados, facilitando un diagnóstico temprano y oportuno de los EIM. Es importante recalcar que estas pruebas pueden ser estandarizadas en cualquier laboratorio convencional porque no requieren reactivos ni equipos sofisticados, inclusive podrían ser candidatas para introducirlas en la tamización neonatal colombiana.
REFERENCIAS BIBLIOGRÁFICAS
1. Trahms C. Inborn errors of metabolism. In: Coulston A, Boushey C. Nutrition in the prevention and treatment of disease. 1st ed. London: Elsevier Academic Press; 2001. p. 209-225. [ Links ]
2. Sanchéz M, Sierra D. Errores Innatos del Metabolismo: aproximación diagnóstica en atención primaria. Bol Pediatr. 2007; 47(200): 111-115. [ Links ]
3. Vela M, Belmont L, Fernández C, Ramírez C, Ibarra I. Frecuencia de enfermedades metabólicas congénitas susceptibles a ser identificadas por el tamiz neonatal. Acta PediatrMex. 2009; 30(3): 156-162. [ Links ]
4. Sanderson S, Green A, Preece MA, Burton H. The incidence of inherited metabolic disorders in the West Midlands, UK. Arch Dis Child. 2006 Nov;91(11):896–9. [ Links ]
5. Wraith JE. Diagnosis and management of inborn errors of metabolism. Arch Dis Child. 1989 Oct;64(10 Spec No):1410–5. [ Links ]
6. Lazzarino G, Amorini AM, Di Pietro V, Tavazzi B. HPLC analysis for the clinical-biochemical diagnosis of inborn errors of metabolism of purines and pyrimidine's. Methods Mol Biol. 2011 Jan;708:99–117. [ Links ]
7. Buist NR. Set of simple side-room urine tests for detection of inborn errors of metabolism. Br Med J. 1968 Jun;2(5607):745–9. [ Links ]
8. Duran M, Gibson M. Simple metabolic screening tests. In: Duran M, Gibson M, editor. Laboratory guide to the methods in biochemical genetics. Heidelberg: Springer; 2008. p. 23–31. [ Links ]
9. Steinmann B, Gitzelmann R, Van den Berghe. Disorders of fructose metabolism. In: Scriver C, Beaudet A, Sly W, et al. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 1489-1520. [ Links ]
10. Ananth N, Praveenkumar GS, Rao KA, Vasanthi, Kakkilaya S. Two cases of hereditary fructose intolerance. Indian J Clin Biochem. 2003 Jul;18(2):87–92. [ Links ]
11. Roe J. A colorimetric method for the determination of fructose in blood and urine. J Biol Chem. 1934; 107: 15-22. [ Links ]
12. Nigam A, Ayagari A. Lab manual in biochemistry, immunology and biotechnology. New Delhi: Tata McGraw-Hill; 2007. [ Links ]
13. Anderson RA, Reddy JM, Oswald C, Zaneveld LJ. Enzymic determination of fructose in seminal plasma by initial rate analysis. Clin Chem. 1979 Oct;25(10):1780–2. [ Links ]
14. Kawasaki T, Akanuma H, Yamanouchi T. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care. 2002 Feb;25(2):353–7. [ Links ]
15. Roe J, Epstein J, Goldstein N. A photometric method for the determination of insulin in plasma and urine. J Biol Chem. 1949 Apr;178(2):839–45. [ Links ]
16. Litwack G. Bioquímica experimental. Barcelona: Ediciones Omega; 1967. [ Links ]
17. Gray DJS. Critical factors in the resorcinol reaction for the determination of fructose. Analyst. 1950;75(891):314-317. [ Links ]
18. Holme E. Disorder of tyrosine degradation. In: Blau N, Duran M, Blaskovics M, Gibson M, editor. Physician's guide to the laboratory diagnosis of metabolic disease. 2nd ed. Heidelberg: Springer; 2003. p. 141–54. [ Links ]
19. Mitchell G, Grompe M, Tanguay R, Lambert M. Hypertyrosinemia. In: Scriver C, Beaudet A, Sly W, et al. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 1777- 1805. [ Links ]
20. Tomoeda K, Awata H, Matsuura T, Matsuda I, Ploechl E, Milovac T, et al. Mutations in the 4-hydroxyphenylpyruvic acid dioxygenase gene are responsible for tyrosinemia type III and hawkinsinuria. Mol Genet Metab. 2000 Nov;71(3):506–10. [ Links ]
21. Kitagawa T. Hepatorenal tyrosinemia. Proc Jpn Acad Ser B Phys Biol Sci. 2012 Jan;88(5):192–200. [ Links ]
22. Couce ML, Aldámiz-Echevarría L, Baldellou A, Blasco J, Bueno MA, Dalmau J, et al. [Recommendations and management of type I hereditary or hepatorenal tyrosinemia]. An Pediatr (Barc). 2010 Nov;73(5):279. [ Links ] e1–4.
23. Ottaway H. A colorimetric method for the estimation of tyrosine. Biochem J. 1958 Feb;68(2):239–44. [ Links ]
24. Knight JA, Robertson G, Wu JT. The chemical basis and specificity of the nitrosonaphthol reaction. Clin Chem. 1983 Nov;29(11):1969–71. [ Links ]
25. Udenfriend S, Weissbach H, Clark C. The estimation of 5-hydroxytryptamine (serotonin) in biological tissues. J Biol Chem. 1955 Jul;215(1):337–44. [ Links ]
26. Udenfriend S, Cooper J. The chemical estimation of tyrosine and tyramine. J Biol Chem. 1952 May;196(1):227–33. [ Links ]
27. Cerrioti G, Spandrio L. Colorimetric determination of tyrosine. Biochem J. 1957 Aug;66(4):607–10. [ Links ]
28. Walter J, Lachmann R, Burgard P. Hyperphenylalaninaemia. In: Saudubray J, Berghe G, Walter J. Inborn metabolic diseases: diagnosis and treatment. 5th ed. Heidelberg: Springer; 2012. p. 251-264. [ Links ]
29. Williams RA, Mamotte CDS, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008 Feb;29(1):31–41. [ Links ]
30. Mundt L, Shanahan K. Graff's textbook of routine urinalysis and body fluids. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2010. [ Links ]
31. Friedemann T, Haugen G. The determination of keto acids in blood and urine. J Biol Chem. 1943 Feb; 147: 415-442. [ Links ]
32. Roe J.Comparative analyses for ascorbic acid by the 2,4-dinitrophenylhydrazine method with the coupling reaction at different temperatures: a procedure for determining specificity. J Biol Chem. 1961 May;236:1611–3. [ Links ]
33. Pavia D, Lampman G, Kriz G, Engel R. Introduction to organic laboratory techniques: A small scale approach. 2nd ed. Belmont: Thomson Books-Cole. 2004. [ Links ]
34. Hassid W, McCready R. Identification of sugar by microscopic appearance of crystalline osazones. Ind Eng Chem Anal. 1942; 14(8): 683-686. [ Links ]
35. Matern D, Rinaldo P. Newborn screening for inherited metabolic disease. In: Hoffmann G, Zschocke J, Nyham W, editor. Inherited metabolic diseases: A clinical approach. Heidelberg: Springer; 2010. p. 251-262. [ Links ]
36. Duran M. Amino acids. In: Nenad B, Duran M, Gibson M, Editor. Laboratory guide to the methods in biochemical genetics. Heiselberg: Springer; 2008. p. 53-89. [ Links ]
37. Salonen K, Niinikoski H, Simell O. Transport defects of amino acids at the cell membrane: cystinuria, lysinuric protein intolerance and Hartnup disorder. In: Saudubray J, Berghe G, Walter J. Inborn metabolic diseases: diagnosis and treatment. 5th ed. Heidelberg: Springer; 2012. p. 363-372. [ Links ]
38. Nakagawa Y, Coe FL. A modified cyanide-nitroprusside method for quantifying urinary cystine concentration that corrects for creatinine interference. Clin Chim Acta. 1999 Nov;289(1-2):57–68. [ Links ]
39. Grote W. A new color reaction for soluble organic sulfur compounds. J Biol Chem. 1931 Jun; 93: 25-30. [ Links ]
40. Wu JT, Wilson LW, Christensen S. Conversion of a qualitative screening test to a quantitative measurement of urinary cystine and homocystine. Ann Clin Lab Sci. 1992 Jan-Feb; 22(1):18–29. [ Links ]
41. Finocchiaro R, D'Eufemia P, Celli M, Zaccagnini M, Viozzi L, Troiani P, et al. Usefulness of cyanide-nitroprusside test in detecting incomplete recessive heterozygotes for cystinuria: a standardized dilution procedure. Urol Res. 1998 Jan;26(6):401–5. [ Links ]
42. Hindmarsh JT. The porphyrias, appropriate test selection. Clin Chim Acta. 2003 Jul 15;333(2):203–7. [ Links ]
43. Zaider E, Bickers DR. Clinical laboratory methods for diagnosis of the porphyrias. Clin Dermatol. 1998 Mar- Apr;16(2):277–93. [ Links ]
44. Gonzáles A. Síntesis y degradación del hemo. Porfiria. Hiperbilirrubinemia. En: Gonzáles A. Principios de bioquímica clínica y patología molecular. Barcelona: Elsevier; 2010. p. 127-137. [ Links ]
45. De Siervi A, Rossetti M V, Parera VE, Mendez M, Varela LS, del C Batlle AM. Acute intermittent porphyria: biochemical and clinical analysis in the Argentinean population. Clin Chim Acta. 1999 Oct;288(1-2):63–71. [ Links ]
46. Méndez M, Rossetti M V, Del C Batlle AM, Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol. 2005 Mar;52(3 Pt 1):417–24. [ Links ]
47. Mauzerall D, Granick S. The occurrence and determination of delta-amino-levulinic acid and porphobilinogen in urine. J Biol Chem. 1956 Mar;219(1):435–46. [ Links ]
48. Lamon J, With TK, Redeker AG. The Hoesch test: bedside screening for urinary porphobilinogen in patients with suspected porphyria. Clin Chem. 1974 Nov;20(11):1438–40. [ Links ]
49. McPherson R, Ben-Ezra J. Basic examination of urine. In: McPherson R, Pincus M. Henry's clinical diagnosis and manahement. 22d ed. Philadelphia: Saunders Elsevier; 2011. p. 446-479. [ Links ]
50. Pierach CA, Cardinal R, Bossenmaier I, Watson CJ. Comparison of the Hoesch and the Watson-Schwartz tests for urinary porphobilinogen. Clin Chem. 1977 Sep;23(9):1666–8. [ Links ]
