










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.28 no.4 Medellín Oct./Dec. 2015
https://doi.org/10.17533/udea.iatreia.v28n4a07
ARTÍCULO DE REVISIÓN
DOI 10.17533/udea.iatreia.v28n4a07
Transición epitelial-mesenquimal en la progresión del adenocarcinoma prostático
Epithelial-mesenquimal transition in the progression of prostatic adenocarcinoma
Transição epitélial-mesenquimal na progressão adenocarcinoma da próstata
Inés Benedetti1; Niradiz Reyes2
1 MD. Patóloga. Grupo de Histopatología, Facultad de Medicina, Universidad de Cartagena, Colombia.
2 PhD. Grupo de Genética y Biología Molecular, Facultad de Medicina, Universidad de Cartagena, Colombia. nreyesr@unicartagena.edu.co
Recibido: septiembre 8 de 2014
Aceptado: enero 28 de 2015
RESUMEN
Mundialmente, el adenocarcinoma prostático es el segundo cáncer diagnosticado en hombres y las metástasis son su principal complicación; se ha descrito la participación en su desarrollo de la transición epitelial-mesenquimal (TEM) proceso fundamental durante el desarrollo embrionario, la remodelación tisular y la cicatrización, que implica pérdida de las propiedades adhesivas y la polaridad epitelial y adquisición del fenotipo mesenquimal que aumenta la movilidad celular individual y permite el desarrollo de características invasivas. Este cambio en el comportamiento celular es mediado por una regulación molecular compleja en la que participa un gran número de vías de señalización, algunas actuando en forma independiente y otras interconectadas; la mayoría converge en el control de la expresión de la E-cadherina, cuya subregulación es el evento molecular clave en este proceso. Diversos estudios señalan una relación estrecha entre la TEM y el desarrollo y progresión de metástasis en carcinomas, pero ha sido menos ampliamente estudiada en el adenocarcinoma prostático. Los objetivos de esta revisión fueron: describir las bases moleculares y morfológicas de este proceso biológico y analizar la influencia de sus reguladores en la adquisición del fenotipo agresivo por las células tumorales, específicamente en lo que tiene que ver con la progresión del adenocarcinoma prostático.
PALABRAS CLAVE
Transición epitelial-mesenquimal, Adhesión Celular, Metástasis, Adenocarcinoma Prostático
SUMMARY
Worldwide, prostate adenocarcinoma is the second most frequently diagnosed cancer in men, and metastases are its most serious complication. The participation in its development of the epithelial-mesenchymal transition (EMT) has been described, a fundamental process during embryonic development, tissue remodeling and wound healing, which involves loss of adhesive properties and epithelial polarity, and acquisition of a mesenchymal phenotype with increasing cellular motility and invasive capability. This change in cellular behavior is mediated by a complex molecular regulation that includes a high number of signalization pathways acting independently or interconnected, many of them converging in the control of E-cadherin expression, whose regulation is the central molecular event of this process. Different studies support a tight link between EMT and progression and metastases development of carcinomas, but it has been less extensively studied in prostate adenocarcinoma. The aim of this review was to describe the molecular and morphological bases of this biological process, and to analyze the participation of regulators in the acquisition of an aggressive phenotype by tumor cells, specifically in regards to prostate adenocarcinoma progression.
KEY WORDS
Epithelial-mesenchymal transition, Cellular Adhesion, Metastasis, Prostate Cancer
RESUMO
Mundialmente, o adenocarcinoma prostático é o segundo câncer diagnosticado em homens e as metástases são sua principal complicação; descreveu-se a participação em seu desenvolvimento da transição epitélio-mesenquimal (TEM) processo fundamental durante o desenvolvimento embrionário, a remodelação tissular e a cicatrização, que implica perda das propriedades adesivas e a polaridade epitelial e aquisição do fenótipo mesenquimal que aumenta a mobilidade celular individual e permite o desenvolvimento de características invasivas. Esta mudança no comportamento celular é mediado por uma regulação molecular complexa na que participa um grande número de vias de sinalização, algumas atuando em forma independente e outras interconectadas; a maioria converge no controle da expressão da Ecadherin, cuja sub-regulação é o evento molecular clave neste processo. Diversos estudos assinalam uma relação estreita entre a TEM e o desenvolvimento e progressão de metástase em carcinomas, mas foi menos amplamente estudada no adenocarcinoma descrever as bases moleculares e morfológicas deste processo biológico e analisar a influência de seus reguladores na aquisição do fenótipo agressivo pelas células tumorais, especificamente em relação com a progressão do adenocarcinoma prostático.
PALAVRAS CHAVES
Transição epitélio-mesenquimal, Adesão Celular, Metástase, Adenocarcinoma Prostático
Cómo citar: Benedetti I, Reyes N. Transición epitelial-mesenquimal en la progresión del adenocarcinoma prostático. Iatreia. 2015 Oct-Dic;28(4):420-33. DOI 10.17533/udea.iatreia.v28n4a07.
INTRODUCCIÓN
Mundialmente, el adenocarcinoma de próstata (CaP) es el segundo cáncer diagnosticado en hombres (1). Como muchos otros tipos de cáncer, en ausencia de metástasis es generalmente tratable (2). Hace solo poco tiempo que se ha comenzado a entender las bases fundamentales de la cascada metastásica, regulada por distintos genes y vías de señalización (3), que comprende una serie de pasos por los cuales las células tumorales hacen lo siguiente: 1) se desprenden y migran más allá del sitio del tumor primario; 2) invaden los tejidos vecinos y penetran a través de la membrana basal; 3) entran a los vasos sanguíneos y/o linfáticos; 4) sobreviven en la circulación; 5) salen de los vasos sanguíneos y/o linfáticos hacia órganos o tejidos distantes; 6) forman nódulos micrometastásicos; y 7) adaptan y reprograman el estroma circundante formando macrometástasis (4).
En la etapa inicial de las metástasis se producen cambios en las células tumorales cuya consecuencia final es su migración a través de la membrana basal con invasión del microambiente circundante, haciendo parte de un proceso caracterizado por pérdida de las uniones intercelulares y reorganización del citoesqueleto, pérdida de la polaridad apical-basal y adquisición de morfología de células fusiformes, todo esto asociado con subregulación del patrón de expresión génica epitelial y aumento de expresión de genes que ayudan a definir el fenotipo mesenquimal (5). Este cambio de las células epiteliales hacia un estado mesenquimal se conoce como transición epitelial-mesenquimal (EMT, por su sigla en inglés), un proceso esencial para el desarrollo embrionario normal, también implicado en la progresión del cáncer hacia un fenotipo invasivo (6), que confiere a las células tumorales una ventaja de supervivencia durante el difícil camino que deben recorrer desde el tumor primario hacia los sitios de metástasis; recientemente han surgido evidencias de la participación de ese proceso en la progresión del CaP (2,7,8).
MECANISMOS DE ADHESIÓN CELULAR
La evolución de los organismos multicelulares permitió el desarrollo de tipos celulares especializados, con diversos fenotipos. Una de las divergencias más tempranas en estos fenotipos fue la distinción entre células epiteliales y células mesenquimales (9,10). Las primeras poseen contactos intercelulares bien desarrollados, están estrechamente unidas, forman una barrera necesaria para la regulación del ambiente interno y mantienen la integridad de los organismos multicelulares (9), mientras que las segundas sostienen las células epiteliales mediante la formación de una matriz extracelular, rara vez forman contactos entre ellas y tienen movilidad (5,11). Otra característica de las células epiteliales son sus dominios de membrana plasmática: el dominio apical expuesto al medio extracelular, la superficie baso-lateral que interactúa con las células vecinas y la membrana basal. El dominio lateral contiene estructuras intercelulares que incluyen uniones estrechas, uniones adherentes y desmosomas, que contribuyen a establecer una polaridad basalapical, además de las llamadas gap junctions, uniones formadoras de canales que permiten la comunicación intercelular (11-13). En cada uno de los tipos de unión, juegan un papel fundamental las glicoproteínas de adhesión transmembrana entre las que se encuentran las cadherinas clásicas pertenecientes a la superfamilia de las cadherinas (como E-cadherina, P-cadherina y N-cadherina), que median la unión célula-célula, y la familia de las integrinas que lo hacen con la unión célula-matriz extracelular (13,14). Las moléculas de cadherinas se unen en forma homotípica en presencia de calcio, mediante interacciones entre sus dominios extracelulares, mientras que los intracelulares se unen a los filamentos del citoesqueleto. Esta unión depende del ensamblaje de un grupo de proteínas intracelulares de anclaje en la cola de las moléculas de cadherinas, principalmente ß-catenina y placoglobina (14-16). La ß-catenina, además, hace parte de la vía de señalización intracelular Wnt, de modo que algunas regiones de su molécula participan en las uniones celulares, y otras, en la regulación de la expresión génica. Así, cuando se desintegran las uniones celulares, la ß-catenina libre migra al núcleo y activa la transcripción de genes diana, y al contrario, componentes de esta vía (que regulan la fosforilación y degradación de la ß-catenina) pueden controlar su disponibilidad para hacer parte de las uniones adherentes (14,15,17).
Otras estructuras unen las células epiteliales a la lámina basal: las uniones matriz-célula enlazadas a actina que anclan los filamentos de esta última a la matriz, y los hemidesmosomas donde la integrina α6ß4 ancla los filamentos de queratina de las células a la laminina de la membrana basal. Igualmente, se organizan complejos de proteínas alrededor de las colas intracelulares de las integrinas produciendo señales que influyen en los procesos de polaridad y migración celular (13). Esta organización de las células epiteliales se pierde durante la fase inicial de la génesis tumoral, que en el caso de los tejidos epiteliales da origen a los carcinomas, el tipo más frecuente (90 %) de neoplasia maligna en humanos (3,18), cuyas células en las etapas avanzadas de la progresión adquieren diferenciación mesenquimal, correlacionada con su potencial metastásico (19).
TRANSICIÓN EPITELIAL-MESENQUIMAL
La transición epitelial-mesenquimal es un proceso biológico en el que las células epiteliales polarizadas que normalmente interactúan con su membrana basal pierden su organización y las uniones intercelulares, reprograman la expresión génica y desarrollan múltiples cambios bioquímicos que las capacitan para adquirir un fenotipo mesenquimal, que incluye cambio en los programas de señalización que definen la forma y organización del citoesqueleto, lo que lleva a una capacidad migratoria aumentada, invasividad, resistencia a la apoptosis y producción elevada de componentes de la matriz extracelular (16,20,21). Continúa con la degradación de la membrana basal y la formación de células mesenquimales que pueden migrar en forma individual, invadir el tejido circundante y desplazarse hasta sitios distantes (21). Los cambios clave en la EMT son: la desestabilización coordinada de las uniones intercelulares y la estimulación de asociaciones dinámicas célula-matriz extracelular requeridas para la locomoción celular (5). El concepto inicial de transformación epitelial-mesenquimal lo introdujo Elizabeth Hay, quien en 1967 se percató de su participación en el desarrollo embrionario (22,23), con evidencias adicionales de una serie de eventos altamente coordinada y específica, que definen la transición entre células epiteliales y mesenquimales (24).
La EMT se inicia con la pérdida de la polaridad apicalbasal a medida que se pierden las uniones estrechas, permitiendo que se entremezclen los componentes de las membranas apical y baso-lateral, y acompañada por disminución de la expresión de claudinas y ocludinas (11). Las uniones intercelulares adherentes comienzan a desensamblarse, la E-cadherina se libera de la membrana y es degradada (25), se separan los desmosomas, se debilitan las uniones tipo gap y por último se degrada la membrana basal (11,15). Las proteínas de la superficie celular como la E-cadherina y las integrinas, que median la conexión entre células vecinas y con la membrana basal, respectivamente, son reemplazadas por N-cadherina e integrinas que proveen a las células de uniones transitorias induciéndolas al fenotipo mesenquimal. Los componentes del citoesqueleto se reorganizan y la actina periférica es reemplazada por fibras de estrés, mientras que los filamentos intermedios de citoqueratina son reemplazados por vimentina (figura 1) (9). El fenotipo mesenquimal resultante se caracteriza por cambios morfológicos y moleculares tales como: adquisición de forma alargada parecida a la de los fibroblastos, sobrerregulación de marcadores mesenquimales (N-cadherina, vimentina, actina de músculo liso) y componentes de la matriz extracelular (colágeno α1 y α2), subregulación de marcadores de superficie de células epiteliales y componentes del citoesqueleto (E-cadherina, claudinas, ocludinas, citoqueratinas), sobrerregulación y/o translocación nuclear de factores de transcripción específicos (Snail, Slug, ZEB1/2, Twist1/2), resistencia a la apoptosis y aumento en la capacidad de invasión y migración a través de la matriz extracelular sin contacto célula-célula (20,21,26,27).
La EMT tiene lugar en tres contextos biológicos distintos, con diferentes consecuencias funcionales: EMT tipo 1 asociada con procesos de desarrollo, EMT tipo 2 asociada con procesos de cicatrización y regeneración tisular, y EMT tipo 3 asociada a progresión tumoral (21). El tipo 1 ocurre durante el desarrollo embrionario, asociado con la formación del embrión y el desarrollo de órganos, generando diversos tipos celulares y células mesenquimales primitivas que también tienen el potencial para llevar a cabo el proceso inverso, o sea, la transición mesenquimal-epitelial (MET, por su sigla en inglés), para generar un epitelio secundario (21). El tipo 2 se asocia con la cicatrización de las heridas, la regeneración tisular y la fibrosis de órganos; se inicia con la formación de fibroblastos y otras células para reconstruir el tejido dañado. Está asociado con inflamación y se detiene cuando esta desaparece, o lleva a fibrosis en caso de inflamación persistente, o a destrucción del órgano (21). El tipo 3 consiste en el cambio de células cancerosas de origen epitelial en células tumorales con características mesenquimales capaces de invadir y diseminarse llevando a la formación de metástasis (20) (figura 2). Estos dos últimos tipos de EMT se asemejan en los factores de crecimiento conductores y en la secreción de proteasas que les permiten a estas células remodelar la matriz extracelular circundante (28).
TRANSICIÓN EPITELIAL-MESENQUIMAL ASOCIADA A PROGRESIÓN TUMORAL Y METÁSTASIS
En su etapa inicial, los carcinomas se caracterizan por aumento en la proliferación de células epiteliales y angiogénesis. La aparición subsecuente de invasividad ocurre al inicio de la etapa final del proceso, que finalmente lleva a la diseminación metastásica (17). Entre los controles genéticos y mecanismos bioquímicos subyacentes a la adquisición de este fenotipo invasivo, se ha propuesto como un mecanismo crítico la activación de un programa de EMT (25). En este contexto no ocurre la inducción coordinada y ordenada de una EMT completa, sino que, en su lugar, las señales ambientales altamente variables, junto con la heterogeneidad genética del tumor, pueden llevar a diversos grados de plasticidad epitelial y a reactivación de programas migratorios asociados con el desarrollo, de lo que resulta la inducción de migración celular individual o colectiva. Esto sugiere que la EMT no es simplemente un mecanismo de diseminación local de las células tumorales a partir de un sitio primario, sino que se induce un programa con las propiedades necesarias para la progresión tumoral (9).
Por otra parte, el hecho de que las células tumorales que migran y se establecen en un sitio distante formando un tumor secundario sean histopatológicamente similares a sus progenitoras en el tumor primario implica que, además del papel facilitador de la EMT en la diseminación tumoral, se requiere que estas células se despojen de su fenotipo mesenquimal por medio de un proceso de transición mesenquimal-epitelial (MET) durante la formación de dicho tumor secundario (3). Esta tendencia a desarrollar la MET probablemente refleje el microambiente local que las células encuentran en el órgano distante y muy posiblemente la ausencia de las señales presentes en el tumor primario que desencadenaron la EMT (17); además, la recuperación del fenotipo epitelial cohesivo es ventajosa en este contexto y por tanto la EMT puede ser un fenómeno transitorio en la progresión tumoral de muchos carcinomas (5).
En relación con los mecanismos que inician la EMT, se ha planteado que las alteraciones genéticas y epigenéticas que sufren las células tumorales durante la formación del tumor primario hacen que respondan a las señales inductoras de EMT provenientes del estroma asociado al tumor (17). La EMT se inicia en un contexto en el que juegan un papel principal los factores de crecimiento y sus receptores, moléculas relacionadas con la matriz extracelular y varias vías de señalización celular (5). En diversos estudios llevados a cabo en líneas celulares, se han identificado numerosos factores de crecimiento como inductores de la EMT incluyendo: el factor de crecimiento transformante ß (TGF-ß), el factor de crecimiento epidérmico (EGF) (29,30), Wnt (31), Snail/Slug (29,32,33), Twist (34) y Six1 (9,35-37). La regulación molecular de la EMT es muy compleja, involucra gran cantidad de vías de señalización actuando en forma independiente o interconectadas, la mayoría convergen en el control de la expresión de E-cadherina, cuya subregulación es el evento molecular clave en este proceso, pues conduce a pérdida de uniones intercelulares y a desestabilización de la arquitectura epitelial (5,6).
EXPRESIÓN DE E-CADHERINA Y TRANSICIÓN EPITELIAL-MESENQUIMAL
La E-cadherina es una glicoproteína transmembrana que regula la adhesión intercelular y la polaridad y forma celulares, al interactuar con moléculas de Ecadherina en las células vecinas y con los filamentos de actina mediante las cateninas (α, ß y p120) (7,13). Es esencial en el mantenimiento de los tejidos epiteliales en los que la fuerza de adhesión de E-cadherina también depende de la interacción citoesqueleto de actina-p120 y la activación de integrinas, lo que sugiere que las cadherinas e integrinas actúan recíprocamente y su represión es un paso crucial en la EMT (23). El control de su expresión durante la EMT y la MET se da en la transcripción y en las vías de señalización que participan en su estabilización. Durante los procesos de desarrollo existen subexpresión y sobreexpresión altamente controladas de diferentes cadherinas, asociadas con ganancia o pérdida de adhesión celular (15). En la mayoría de los carcinomas la progresión tumoral se asocia con pérdida de expresión y/o función de la E-cadherina por las células tumorales, cuya conexión con la EMT se ha establecido en muchos estudios, que además la han relacionado con invasividad, metástasis y mal pronóstico (30,38).
En el CaP se ha reportado asociación entre tumores con bajo índice de Gleason y patrón normal de Ecadherina, y su expresión reducida en tumores avanzados y con alto índice de Gleason, por lo que se la propone como un posible indicador de pronóstico (19,31,32,38). En estos tumores, a semejanza de lo observado en algunos procesos de desarrollo, hay neoexpresión de N-cadherina y expresión aberrante de otros tipos de cadherinas, lo que se asocia con mayor capacidad de invasión, debido a que las interacciones homotípicas entre N-cadherinas son más débiles que las interacciones entre E-cadherinas (15,16,33). En líneas celulares de CaP se ha informado esta alteración en el patrón de expresión de cadherinas, asociada con la transición de adenocarcinoma andrógeno-dependiente a un estado de independencia de andrógenos (30,34,39,40); igualmente, en tejidos con CaP de alto grado y metastásico, en los que se encontró expresión elevada de N-cadherina y cadherina 11, ausentes en el tejido prostático sano, por lo que se propone que las células con estas alteraciones adquieren una ventaja selectiva para la invasión (19,35,40). El aumento de expresión de N-cadherina también se ha asociado a privación androgénica en líneas celulares y en adenocarcinoma prostático (36).
Durante la progresión tumoral, diversos mecanismos pueden inactivar funcionalmente la E-cadherina; entre ellos se incluyen: mutaciones somáticas, que son raras excepto en los carcinomas de tipo difuso que albergan una mutación de inactivación del gen de la E-cadherina (5,31); disminución en la expresión génica por metilación del promotor, que parece ser el mecanismo más frecuente (37), y represión transcripcional (15,41).También se ha descrito una regulación postraduccional, por degradación proteolítica de la molécula original de E-cadherina que lleva a una forma truncada inactiva en la que se ha removido el dominio de ß-catenina (38,42). Por otro lado, la reexpresión de E-cadherina encontrada en la mayoría de las células tumorales de CaP metastásico a ganglio linfático, cuyo tumor primario mostraba subregulación de la misma, ha llevado a plantear que este mecanismo facilita el establecimiento en sitios distantes y el crecimiento de las metástasis, y hace poco probable que la inactivación genética irreversible por mutaciones o pérdida alélica a nivel de 16q2.3 sea causa de esta pérdida de expresión de E-cadherina en el adenocarcinoma prostático (31,38). Como consecuencia de dicha reexpresión ectópica, se reorganizan los complejos de adhesión celular epitelial y se suprime la proliferación celular haciendo que estas células pierdan su fenotipo mesenquimal (19,28).
REGULACIÓN POR FACTORES DE TRANSCRIPCIÓN
Los cambios en la expresión génica que llevan a represión del fenotipo epitelial y activación del mesenquimal son coordinados por un grupo de reguladores principales, los factores de transcripción: Snail y Slug; ZEB: ΔEF1/ZEB1 y Sip1/ZEB2 /SIP1/ZEB2; factores bHLH como E47 y Twist (16,23,37,43) (figura 3). Estos represores, cuya expresión es activada tempranamente en la EMT, se unen a las cajas E proximales del promotor de E-cadherina; la mayor afinidad la tiene Snail, cuya actividad está muy regulada: produce incremento de Sip1/ZEB2, mientras que TGF-ß induce la expresión de Snail y Slug (21,44). Snail es un factor de transcripción que media la EMT por subregulación de moléculas de adhesión como E-cadherina, claudina y ocludina, e incremento de marcadores mesenquimales como vimentina, fibronectina y metaloproteinasas. Su efecto es aumentar la migración y la invasión, lo que refuerza el papel central que juega la pérdida de la E-cadherina en el proceso de la EMT (21,29,44) que, a su vez, promueve la señalización de Wnt y se asocia con un nivel alto de Snail en el núcleo (21,45). La actividad de Snail es controlada por la acción de varias quinasas, y su fosforilación por la glucógeno-sintasaquinasa (GSK-3ß) promueve su traslado del núcleo al citosol donde es degradado, mientras que su fosforilación por la quinasa activada p21 (PAK1) y la proteína quinasa D1 (PKD1) induce su acumulación en el núcleo donde tiene función represora (7,29).
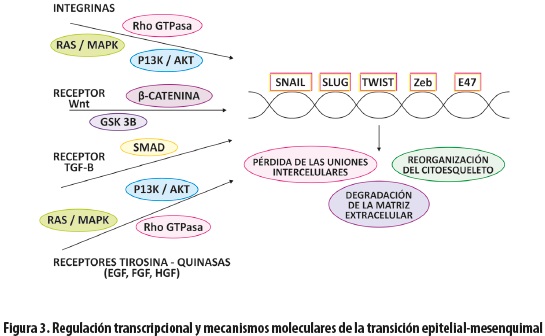
Otro mecanismo reportado para la inactivación de E-cadherina es el silenciamiento epigenético mediante la metilación de su promotor, observada en el CaP. Esto ocurre como un evento temprano en la carcinogénesis, de modo que las células con alelos metilados pueden ser responsables del inicio del proceso metastásico, y posteriormente la disminución en la metilación y la reexpresión del gen pueden contribuir a la supervivencia celular (15). En ocho tipos de carcinoma, incluyendo el CaP, se ha encontrado metilación aberrante del ADN en una gran isla CpG en el extremo 5' del promotor de E-cadherina lo cual se correlaciona con disminución de su expresión (37). Además, la represión inducida por Snail, Slug, ZEB1 y SIP1/ZEB2 involucra el reclutamiento de un complejo represor, lo que lleva a un patrón compacto de cromatina, lo que a su vez establece una conexión entre la represión transcripcional y los mecanismos epigenéticos en la inducción de EMT (15,37,46,47). La regulación de la expresión de E-cadherina es similar durante los procesos de desarrollo y la progresión tumoral, con una participación jerárquica y específica de los diferentes factores en la represión de su expresión durante la invasión (15,43). Así, la expresión transitoria de Snail o Sip1/ZEB2 puede estar involucrada en la inducción del proceso de invasión, mientras que Slug, ZEB y E47 lo estarían en el mantenimiento del fenotipo invasivo, con diferentes patrones de expresión de estos represores en los distintos estadios de la progresión tumoral (15).
En el CaP se ha demostrado la inducción de EMT por factores de crecimiento mediante aumento de la expresión o activación de Snail: TGF-ß y EGF promueven su localización nuclear en las células PC3, y lo mismo hace VEGF-A cuando se une a su receptor neuropilina-1. Este además participa en la inducción de la EMT en respuesta a la hipoxia (48,49). También se ha encontrado sobreexpresión de Snail en el CaP, con aumento progresivo desde el tejido benigno hasta las metástasis, asociado con índice alto de Gleason y mal pronóstico (29,32,50).
Wnt-ß-CATENINA EN LA TRANSICIÓN EPITELIALMESENQUIMAL
La vía de señalización Wnt/ß-catenina establece una conexión entre adhesión y respuesta celulares dirigida por transcripción génica, pues al compartir la molécula de ß-catenina, la adhesión mediada por E-cadherina y la vía Wnt/ß-catenina dependen la una de la otra (5). Esta vía está implicada en la EMT que ocurre durante el desarrollo y también durante la progresión del cáncer; en estudios in vitro se confirma que puede inducir plasticidad epitelial y sobrerregular el marcador mesenquimal vimentina (9). La ß-catenina se encuentra en tres compartimientos subcelulares diferentes: la membrana celular, el citosol y el núcleo. En ausencia de la señalización mediada por Wnt, gran parte de ella es secuestrada por E-cadherina en las uniones adherentes y estrechas para mantener el fenotipo epitelial al prevenir su participación como factor de transcripción para la EMT. Simultáneamente, la fracción de ß-catenina presente en el citosol es fosforilada rápidamente por GSK-3ß en el complejo APC/GSK-3ß/axin, lo que permite su degradación subsiguiente por el proteasoma (51). Cuando está activada la señalización por Wnt, se encuentra inhibida la GSK-3ß, con lo cual la ß-catenina no puede ser fosforilada y degradada, lo que lleva a su acumulación en el citosol, translocación al núcleo y unión a factores de transcripción que activan genes blanco que promueven la proliferación de las células cancerosas e inducen la EMT (5,7,23).
Se puede, además, inducir la señalización por Wnt/ß- catenina ya sea por subregulación de E-cadherina, lo que aumenta la ß-catenina citoplasmática, o por activación de la vía PI3K/Akt, o de quinasa ligada a integrina (ILK) que inhibe GSK-3ß y MAPK/Erk que pueden unírsele e inactivarla (5,52,53). Por lo tanto, el secuestro de ß-catenina en el citoplasma es importante en la preservación de las características epiteliales de las células tumorales (54), mientras que su movimiento al núcleo se asocia con pérdida de expresión de E-cadherina, y con la susceptibilidad a entrar en EMT, por aumento de la transcripción de genes asociados con proliferación celular (17,55,56). En el CaP se ha reportado esta disminución de ß-catenina citoplasmática y su aumento en el núcleo (17,41). La disminución de expresión de E-cadherina en las células del adenocarcinoma prostático lleva a redistribución de la ß-catenina citoplasmática hacia la membrana y a reducción en la transcripción mediada por el receptor de andrógenos (17,57,58). La subregulación de los complejos E-cadherina/ß-catenina se correlaciona en forma significativa con altos índices de Gleason, y en estos casos se encuentra metilación del promotor de E-cadherina, lo que sumado a su detección en la neoplasia intraepitelial prostática (PIN, por su sigla en inglés) sugiere un papel significativo en la carcinogénesis y la progresión del CaP (17,41). Además, se ha encontrado asociación entre la oncogénesis prostática generada por fusión de los genes TMPRSS2-ERG y la vía Wnt, con inducción de EMT por aumento de actividad del gen relacionado con los factores de transcripción ETS (ERG) que causa sobreexpresión de Snail (59).
TGF-ß EN LA TRANSICIÓN EPITELIAL-MESENQUIMAL
TGF-ß es una citocina de expresión ubicua cuya unión a las células diana se hace mediante receptores I y II, e inicia múltiples cascadas de señalización. Induce la EMT en procesos asociados al desarrollo, la cicatrización y la progresión tumoral (16). Como resultado del estímulo con TGF-ß se producen pérdida de la polaridad apical-basal, subregulación de las uniones célulacélula, expresión de proteínas mesenquimales como vimentina en el citoesqueleto, además de proteasas extracelulares, lo que lo hace uno de los más potentes inductores de EMT, actividad que depende del contexto, pues tiene también un efecto paradójico como supresor de tumor (9,60). Su vía de señalización es mediada por el receptor TGF-ß tipo I que fosforila y activa los factores Smad que regulan la transcripción de genes diana específicos, como Snail y Slug. Otros reguladores de EMT, como ZEB1, SIP1/ZEB2, E12/E47 y Twist1, también pueden ser regulados por TGF-ß (61). Además, puede señalizar en forma independiente de Smad por activación de (i) MAP quinasas: ERK1/2, p38MAPK (5,53) y JNK; (ii) mediadores de supervivencia celular: PI3K, AKT1/2 y Mtor; (iii) mediadores de inflamación: NF-κB, Cox-2 y prostaglandinas; (iv) proteínas pequeñas de unión a GTP: Ras, RhoA, Rac1 y Cdc42; y (v) tirosina-quinasas no receptoras: Src y FAK (62). Se ha reportado que las células de cáncer de próstata sobreexpresan TGF-ß1, que es capaz de inducir EMT junto con el EGF, por subregulación de E-cadherina y sobrerregulación de vimentina, Snail y ligando de unión al receptor NFKB activado (RANKL); este último se expresa normalmente en los osteoblastos, estimula la maduración de los osteoclastos y la resorción ósea, por lo que es posible que la EMT, mediante aumento de expresión de RANKL, promueva el aumento del recambio óseo y la subsecuente colonización del hueso durante la formación de las metástasis del CaP (49).
RECEPTORES TIROSINA-QUINASA EN LA TRANSICIÓN EPITELIAL-MESENQUIMAL
Los factores de crecimiento que actúan a través de receptores tirosina-quinasa pueden inducir la EMT parcial o completa (16,51). Las vías de EMT en respuesta al estímulo de estos receptores involucran Ras/MAPK, PI3K/Akt, Rho/Rac o Src (28); en la EMT inducida por estos receptores es esencial la señalización por medio de Akt. La activación de la vía ERK-MAPK es inducida además por las mutaciones en los genes Ras y Raf en las células cancerosas y también contribuye a la EMT, pues Ras o Raf inducen la expresión de Snail y Slug, represores directos de E-cadherina e inductores de EMT (5,16). Además, Akt sobreexpresa Snail y fosforila e inhibe GSK-3ß, lo que lleva a incremento en la señalización por ß-catenina y Snail y promueve la EMT (5,7,27). El cambio de fenotipo epitelial a mesenquimal se ha asociado con el control de la familia de GTP-asas pequeñas: Rho, Rac y Cdc42, que enlazan la activación del receptor de superficie con el esqueleto de actina, afectan corriente abajo las MAPK, alteran la transcripción génica y moldean el fenotipo celular durante la EMT (9). Por su parte, PI3K activa Rho, involucrada junto con Rac en la adhesión celular mediada por E-cadherina y en el control del equilibrio entre el ensamble y el desensamble de actina, responsable de la forma y motilidad celulares; la EMT inducida por TGF-ß depende de Rho y se acompaña de activación de la vía PI3K/Akt y aumento de expresión de N-cadherina (5,27).
El factor de crecimiento de fibroblastos (FGF), el factor de crecimiento de hepatocitos (HGF) y el factor de crecimiento epidérmico (EGF) inducen la EMT acompañada de aumento de Snail y Slug. El factor de crecimiento derivado de plaquetas (PDGF) induce disolución de uniones adherentes y represión de E-cadherina en células de adenocarcinoma de colon (16). En estos procesos se ha demostrado además la participación de DAB2IP, otro miembro de la familia de proteínas de activación de GTP-asas Ras, que facilita la activación de GSK-3ß, lo que disminuye la acumulación nuclear de ß-catenina y su actividad transcripcional; este proceso ha sido descrito en líneas celulares de CaP y llevó a proponer a DAB2IP como supresor de la EMT, cuya subregulación puede aumentar la capacidad de hacer metástasis a ganglios linfáticos, asociada con hiperactivación de la vía Wnt/ß-catenina, y su pérdida lleva a CaP agresivo (63).
MATRIZ EXTRACELULAR Y SEÑALIZACIÓN DE INTEGRINAS
Las integrinas son las principales moléculas en la unión célula-matriz extracelular y permiten a las células recibir señales de la matriz mediante moléculas de señalización (5). Su expresión cambia durante la transición de célula epitelial a mesenquimal: subregulación de α6ß4 que media el contacto con la membrana basal, aumento de la adhesión celular a fibronectina y promoción de la migración celular. Además, el aumento de la expresión de α1ß1 o α2ß1 y su interacción con colágeno tipo I facilitan la degradación de E-cadherina y la translocación nuclear de ß-catenina. Esto se correlaciona con aumento de la expresión de proteasas y remodelación de la matriz extracelular, proceso en el que se pueden liberar factores de crecimiento almacenados como TGF-ß (16). Durante la EMT las células tumorales establecen contacto con la matriz subyacente extendiendo seudópodos y migran por etapas alternantes de adhesión y separación acopladas con la contracción celular. En este proceso participan las proteasas de la matriz: crean rutas de migración para las células tumorales mediante la ruptura de la membrana basal y la matriz intersticial, modifican el microambiente extracelular, liberan y activan factores de movilidad y supervivencia, degradan el dominio extracelular de E-cadherina y liberan y relocalizan la ß-catenina en el citoplasma (5). La mayoría de las proteasas involucradas pertenecen a la familia de las metaloproteinasas de matriz (MMP), de las cuales MMP9 y MMP2 juegan un papel fundamental en la invasión tumoral (5). En el CaP se encontró correlación positiva entre la expresión aumentada de Snail y la expresión o activación de MMP, y se plantea que estas enzimas pueden ser responsables de la pérdida de Ecadherina y el aumento de expresión de N-cadherina mediados por Snail (50). La liberación de citocinas inflamatorias por las células inmunes, los fibroblastos tumorales y el endotelio también induce la EMT (64).
REGULACIÓN POR MICRO-RNA
La sobrerregulación o subregulación de varios micro- RNA es fundamental en la regulación de la EMT (37). Se ha propuesto que la familia miR200 junto con miR205 son marcadores epiteliales y supresores de la EMT (37,65,66); en la EMT inducida por TGF-ß son reprimidos cinco de sus miembros, cuya función normal es reforzar el fenotipo epitelial y regular negativamente el proceso por subregulación de los represores de E-cadherina, ZEB1 y Sip1/ZEB2. A su vez, ZEB1 reprime la expresión de esta familia miR200 creando retroalimentación negativa que induce la progresión de la EMT (61,62). Una retroalimentación negativa similar existe entre ZEB y miR34, Snail y miR34, miR203 y Snail; además, miR34 y miR200c son activados por p53 reprimiendo la EMT (61,62). Let7 también ha sido implicado en la EMT (21,65), al igual que miR10b y Let-7i, regulados por Twist (61,66). En el CaP se ha informado inhibición de la expresión de MMP2 por miR29b, propuesto como participante en el proceso metastásico al estar inhibido en la línea celular altamente metastásica PC3 y en tejido tumoral. También se ha descrito represión de Snail por miR29b y mir30, de manera que su expresión aumentada puede revertir la EMT (67), mientras que miR143 y miR145 inhiben la migración de las células del CaP por inducción de E-cadherina (68). En estas mismas células transfectadas con miR203 se observó aumento de E-cadherina en la membrana y disminución de SIP1/ZEB2, vimentina y fibronectina (69).
APLICACIÓN CLÍNICA
Actualmente es posible determinar en el laboratorio de patología, mediante inmunohistoquímica, la expresión de estas proteínas involucradas en la EMT en el CaP. Varios estudios han evaluado sus niveles de expresión y algunos han encontrado correlación entre la expresión inmunohistoquímica de E-cadherina y N-cadherina y los niveles del antígeno prostático específico (PSA), el índice de Gleason y la presencia de invasión y metástasis (23,31,36,70). Otros informan diferencia en el patrón de expresión de E-cadherina entre las micro y macrometástasis ganglionares lo cual puede ser evidencia del proceso de MET previamente descrito, indicando un mayor potencial metastásico, de modo que la evaluación de su expresión tendría implicaciones terapéuticas potenciales (71). También se ha informado pérdida de E-cadherina, aumento de Snail en el núcleo y sobreexpresión de vimentina en el citoplasma de las células tumorales, asociados con alto índice de Gleason y mal pronóstico; sin embargo, solo E-cadherina estuvo asociada con muerte por CaP (32). Otro trabajo reciente halló disminución significativa de la expresión de E-cadherina en el CaP comparado con el tejido adyacente, principalmente en el citoplasma, y se ha planteado la inhibición de la formación del complejo E-cadherina/ß-catenina. Por su parte, N-cadherina tuvo una mayor expresión en el tejido con cáncer, lo que se cataloga como un Cadherin- switch en el CaP, similar al descrito previamente por otros autores (17,19,35). TGF-ß y Twist1 también mostraron mayor expresión en el tejido con cáncer, inversamente proporcional a la diferenciación del tumor, y tanto su aumento como el de la N-cadherina se correlaciona con niveles más altos de PSA y metástasis óseas (72). Además, se ha estudiado su relación con la evolución después del tratamiento, y se ha encontrado asociación entre la pérdida de E-cadherina en el RNA mensajero y la mala respuesta en pacientes sometidos a tratamiento no quirúrgico (73). La expresión aumentada de N-cadherina en un porcentaje considerable de casos de CaP, mayor en los tumores resistentes a la castración y asociada con pérdida o disminución de la expresión del receptor de andrógenos, plantea una posible regulación sobre este y sugiere una ventaja de crecimiento de estas células sobre las N-cadherina negativas (74). Esto ha llevado a proponer la N-cadherina como posible blanco terapéutico, con una relación inversa entre la EMT y la señalización androgénica en el CaP (2,74). Algunos autores especulan sobre una posible relación entre la expresión de N-cadherina y la adhesión a estructuras neurales que la expresan, que podría conducir a invasión perineural, una vía conocida de diseminación del CaP asociada a mal pronóstico (2).
CONCLUSIONES
La progresión de los carcinomas implica una serie de alteraciones en las que múltiples moléculas actúan interconectadas, reguladas por vías de señalización que pueden converger hacia el cambio fenotípico de las células epiteliales tumorales a un linaje mesenquimal con características migratorias e invasivas, que les permiten diseminarse y establecerse en sitios distantes formando metástasis. Este proceso descrito como transición epitelial mesenquimal tiene una participación crucial en la progresión tumoral, la invasión y las metástasis. Se han encontrado evidencias en favor de su participación en la progresión del CaP, como la presencia del cadherin-switching en las células tumorales, y expresión de marcadores asociados como Snail, Twist1 y TFG-ß; este es un potente inductor de la EMT por medio de cambios en la expresión génica y la activación de factores de transcripción.
REFERENCIAS BIBLIOGRÁFICAS
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014 Jan-Feb;64(1):9-29. [ Links ]
2. Armstrong AJ, Freedland SJ, Garcia-Blanco M. Epithelial- mesenchymal transition in prostate cancer: providing new targets for therapy. Asian J Androl. 2011 Mar;13(2):179-80. [ Links ]
3. Yao D, Dai C, Peng S. Mechanism of the mesenchymal- epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011 Dec;9(12):1608-20. [ Links ]
4. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006 Aug;12(8):895-904. [ Links ]
5. Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology. 2007 Jun;39(3):305-18. [ Links ]
6. Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008 Feb;68(3):937-45. [ Links ]
7. Deep G, Jain AK, Ramteke A, Ting H, Vijendra KC, Gangar SC, et al. SNAI1 is critical for the aggressiveness of prostate cancer cells with low E-cadherin. Mol Cancer. 2014 Feb;13:37. [ Links ]
8. Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci. 2013 Jan;126(Pt 1):21-9. [ Links ]
9. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010 Jun;15(2):117-34. [ Links ]
10. Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn. 2005 Jul;233(3):706-20. [ Links ]
11. Huang RY, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci. 2012 Oct;125(Pt 19):4417-22. [ Links ]
12. Feigin ME, Muthuswamy SK. Polarity proteins regulate mammalian cell-cell junctions and cancer pathogenesis. Curr Opin Cell Biol. 2009 Oct;21(5):694-700. [ Links ]
13. Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996 Feb;84(3):345-57. [ Links ]
14. Kemler R. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993 Sep;9(9):317-21. [ Links ]
15. Peinado H, Portillo F, Cano A. Transcriptional regulation of cadherins during development and carcinogenesis. Int J Dev Biol. 2004;48(5-6):365-75. [ Links ]
16. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014 Mar;15(3):178-96. [ Links ]
17. Jaggi M, Johansson SL, Baker JJ, Smith LM, Galich A, Balaji KC. Aberrant expression of E-cadherin and beta-catenin in human prostate cancer. Urol Oncol. 2005 Nov-Dec;23(6):402-6. [ Links ]
18. Fidler IJ. Critical determinants of metastasis. Semin Cancer Biol. 2002 Apr;12(2):89-96. [ Links ]
19. Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET, Jansen CF, Bussemakers MJ, et al. Cadherin switching in human prostate cancer progression. Cancer Res. 2000 Jul;60(13):3650-4. [ Links ]
20. Tiwari N, Gheldof A, Tatari M, Christofori G. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol. 2012 Jun;22(3):194-207. [ Links ]
21. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009 Jun;119(6):1420-8. [ Links ]
22. Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel). 1995;154(1):8-20. [ Links ]
23. Acloque H, Thiery JP, Nieto MA. The physiology and pathology of the EMT. Meeting on the epithelial-mesenchymal transition. EMBO Rep. 2008 Apr;9(4):322-6. [ Links ]
24. Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev. 2003 Nov;120(11):1351-83. [ Links ]
25. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009 Jun;28(1-2):15-33. [ Links ]
26. Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006 Mar;172(7):973-81. [ Links ]
27. Zhu QC, Gao RY, Wu W, Qin HL. Epithelial-mesenchymal transition and its role in the pathogenesis of colorectal cancer. Asian Pac J Cancer Prev. 2013;14(5):2689-98. [ Links ]
28. Leopold PL, Vincent J, Wang H. A comparison of epithelial-to-mesenchymal transition and re-epithelialization. Semin Cancer Biol. 2012 Oct;22(5-6):471-83. [ Links ]
29. Smith BN, Odero-Marah VA. The role of Snail in prostate cancer. Cell Adh Migr. 2012 Sep-Oct;6(5):433-41. [ Links ]
30. Murali AK, Norris JS. Differential expression of epithelial and mesenchymal proteins in a panel of prostate cancer cell lines. J Urol. 2012 Aug;188(2):632-8. [ Links ]
31. De Marzo AM, Knudsen B, Chan-Tack K, Epstein JI. Ecadherin expression as a marker of tumor aggressiveness in routinely processed radical prostatectomy specimens. Urology. 1999 Apr;53(4):707-13. [ Links ]
32. Whiteland H, Spencer-Harty S, Thomas DH, Davies C, Morgan C, Kynaston H, et al. Putative prognostic epithelial-to-mesenchymal transition biomarkers for aggressive prostate cancer. Exp Mol Pathol. 2013 Oct;95(2):220-6. [ Links ]
33. Sandig M, Voura EB, Kalnins VI, Siu CH. Role of cadherins in the transendothelial migration of melanoma cells in culture. Cell Motil Cytoskeleton. 1997;38(4):351-64. [ Links ]
34. Tran NL, Nagle RB, Cress AE, Heimark RL. N-Cadherin expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion withStromal cells. Am J Pathol. 1999 Sep;155(3):787-98. [ Links ]
35. Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007 Dec;13(23):7003-11. [ Links ]
36. Jennbacken K, Tesan T, Wang W, Gustavsson H, Damber JE, Welén K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr Relat Cancer. 2010 May;17(2):469-79. [ Links ]
37. Wang Y, Shang Y. Epigenetic control of epithelial-tomesenchymal transition and cancer metastasis. Exp Cell Res. 2013 Jan;319(2):160-9. [ Links ]
38. Rubin MA, Mucci NR, Figurski J, Fecko A, Pienta KJ, Day ML. E-cadherin expression in prostate cancer: a broad survey using high-density tissue microarray technology. Hum Pathol. 2001 Jul;32(7):690-7. [ Links ]
39. Jennbacken K, Gustavsson H, Welén K, Vallbo C, Damber JE. Prostate cancer progression into androgen independency is associated with alterations in cell adhesion and invasivity. Prostate. 2006 Nov;66(15):1631-40. [ Links ]
40. Bussemakers MJ, Van Bokhoven A, Tomita K, Jansen CF, Schalken JA. Complex cadherin expression in human prostate cancer cells. Int J Cancer. 2000 Feb;85(3):446-50. [ Links ]
41. Kallakury BV, Sheehan CE, Winn-Deen E, Oliver J, Fisher HA, Kaufman RP Jr, et al. Decreased expression of catenins (alpha and beta), p120 CTN, and E-cadherin cell adhesion proteins and E-cadherin gene promoter methylation in prostatic adenocarcinomas. Cancer. 2001 Dec;92(11):2786-95. [ Links ]
42. Vallorosi CJ, Day KC, Zhao X, Rashid MG, Rubin MA, Johnson KR, et al. Truncation of the beta-catenin binding domain of E-cadherin precedes epithelial apoptosis during prostate and mammary involution. J Biol Chem. 2000 Feb;275(5):3328-34. [ Links ]
43. Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000 Feb;2(2):76-83. [ Links ]
44. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. 2008 Nov;19(11):4875-87. [ Links ]
45. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001 Jun;7(6):1267-78. [ Links ]
46. Le Bras GF, Taubenslag KJ, Andl CD. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh Migr. 2012 Jul-Aug;6(4):365-73. [ Links ]
47. Stadler SC, Allis CD. Linking epithelial-to-mesenchymal- transition and epigenetic modifications. Semin Cancer Biol. 2012 Oct;22(5-6):404-10. [ Links ]
48. Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, et al. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010 Apr;17(4):319-32. [ Links ] Erratum in: Cancer Cell. 2010 Jun;17(6):622.
49. Odero-Marah VA, Wang R, Chu G, Zayzafoon M, Xu J, Shi C, et al. Receptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008 Aug;18(8):858-70. [ Links ]
50. Poblete CE, Fulla J, Gallardo M, Muñoz V, Castellón EA, Gallegos I, et al. Increased SNAIL expression and low syndecan levels are associated with high Gleason grade in prostate cancer. Int J Oncol. 2014 Mar;44(3):647-54. [ Links ]
51. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003 Dec;112(12):1776-84. [ Links ]
52. Nelson WJ, Nusse R. Convergence of Wnt, betacatenin, and cadherin pathways. Science. 2004 Mar;303(5663):1483-7. [ Links ]
53. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006 Feb;7(2):131-42. [ Links ]
54. Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol. 2001 May;153(5):1049-60. [ Links ]
55. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002 Jun;2(6):442-54. [ Links ]
56. Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26(5):463-76. [ Links ]
57. Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, et al. Linking beta-catenin to androgen-signaling pathway. J Biol Chem. 2002 Mar;277(13):11336-44. [ Links ]
58. Schweizer L, Rizzo CA, Spires TE, Platero JS, Wu Q, Lin TA, et al. The androgen receptor can signal through Wnt/beta-Catenin in prostate cancer cells as an adaptation mechanism to castration levels of androgens. BMC Cell Biol. 2008 Jan;9:4. [ Links ]
59. Gupta S, Iljin K, Sara H, Mpindi JP, Mirtti T, Vainio P, et al. FZD4 as a mediator of ERG oncogene-induced WNT signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2010 Sep;70(17):6735-45. [ Links ]
60. Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGF-ß and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle. 2010 Jun;9(12):2363-74. [ Links ]
61. Wu CY, Tsai YP, Wu MZ, Teng SC, Wu KJ. Epigenetic reprogramming and post-transcriptional regulation during the epithelial-mesenchymal transition. Trends Genet. 2012 Sep;28(9):454-63. [ Links ]
62. Moes M, Le Béchec A, Crespo I, Laurini C, Halavatyi A, Vetter G, et al. A novel network integrating a miRNA-203/SNAI1 feedback loop which regulates epithelial to mesenchymal transition. PLoS One. 2012;7(4):e35440. [ Links ]
63. Xie D, Gore C, Liu J, Pong RC, Mason R, Hao G, et al. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc Natl Acad Sci U S A. 2010 Feb;107(6):2485-90. [ Links ]
64. Jing Y, Han Z, Zhang S, Liu Y, Wei L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011 Aug;1:29. [ Links ]
65. Marie-Egyptienne DT, Lohse I, Hill RP. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: potential role of hypoxia. Cancer Lett. 2013 Nov;341(1):63-72. [ Links ]
66. Zhang J, Ma L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012 Dec;31(3-4):653-62. [ Links ]
67. Ru P, Steele R, Newhall P, Phillips NJ, Toth K, Ray RB. miRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol Cancer Ther. 2012 May;11(5):1166-73. [ Links ]
68. Peng X, Guo W, Liu T, Wang X, Tu X, Xiong D, et al. Identification of miRs-143 and -145 that is associated with bone metastasis of prostate cancer and involved in the regulation of EMT. PLoS One. 2011;6(5):e20341. [ Links ]
69. Saini S, Majid S, Yamamura S, Tabatabai L, Suh SO, Shahryari V, et al. Regulatory Role of mir-203 in Prostate Cancer Progression and Metastasis. Clin Cancer Res. 2011 Aug;17(16):5287-98. [ Links ]
70. Liu Y, Chen XG, Liang CZ. [Expressions of E-cadherin and N-cadherin in prostate cancer and their implications]. Zhonghua Nan Ke Xue. 2014 Sep;20(9):781-6. [ Links ] Chinese.
71. Isebaert S, Haustermans K, Van den Bergh L, Joniau S, Dirix P, Oyen R, et al. Identification and characterization of nodal metastases in prostate cancer patients at high risk for lymph node involvement. Acta Oncol. 2013 Oct;52(7):1336-44. [ Links ]
72. Liu GL, Yang HJ, Liu T, Lin YZ. Expression and significance of E-cadherin, N-cadherin, transforming growth factor-ß1 and Twist in prostate cancer. Asian Pac J Trop Med. 2014 Jan;7(1):76-82. [ Links ]
73. Kachroo N, Warren AY, Gnanapragasam VJ. Multitranscript profiling in archival diagnostic prostate cancer needle biopsies to evaluate biomarkers in non-surgically treated men. BMC Cancer. 2014 Sep;14:673. [ Links ]
74. Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 2010 Dec;16(12):1414-20. [ Links ]
