










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.29 no.2 Medellín Apr./June 2016
https://doi.org/10.17533/udea.iatreia.v29n2a08
DOI 10.17533/udea.iatreia.v29n2a08
ARTÍCULO DE REVISIÓN
Esclerosis lateral amiotrófica: actualización
Amyotrophic lateral sclerosis: update
La esclerose lateral amiotrófica: atualização
Carlos Hugo Zapata-Zapata1; Edwing Franco-Dáger1; Juan Marcos Solano-Atehortúa2; Luisa Fernanda Ahunca-Velásquez3
1 Residente de Neurología, Facultad de Medicina, Universidad de Antioquia, Medellín Colombia. zcarloshz@yahoo.es
2 Profesor de Neurología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
3 Profesora de Cátedra. Instituto de Investigación, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
Recibido: abril 10 de 2015
Aceptado: agosto 12 de 2015
RESUMEN
La esclerosis lateral amiotrófica es una enfermedad neurodegenerativa que tiene consecuencias devastadoras para el paciente y su familia. Aún no existe claridad sobre su etiología, cerca del 10 % de los pacientes tienen patrón hereditario. La prevalencia mundial varía entre 2 y 11 casos por 100.000 habitantes; el rango de edad de presentación es de 58 a 63 años para los casos esporádicos, y de 47 a 52 años para los familiares, con una ligera predilección por el sexo masculino. Las manifestaciones clínicas incluyen signos de daño de las neuronas motoras superior e inferior tanto en las extremidades como en la musculatura bulbar, y en algunos pacientes hay deterioro cognitivo frontotemporal. El diagnóstico continúa siendo fundamentalmente clínico, apoyado por estudios neurofisiológicos; de estos, la electromiografía de aguja ha sido el más útil para el diagnóstico temprano. No existe tratamiento curativo y solo un medicamento, el riluzol, ha demostrado efectividad para retrasar el uso de ventilación mecánica y prolongar levemente la supervivencia. Por tanto, el tratamiento de estos pacientes se basa en medidas de soporte, especialmente en los aspectos de nutrición y ventilación, además de controlar los síntomas motores y no motores de la enfermedad.
PALABRAS CLAVE
Diagnóstico, Enfermedad de la Neurona Motora, Epidemiología, Esclerosis Amiotrófica Lateral, Signos y Síntomas, Terapéutica
SUMMARY
Amyotrophic lateral sclerosis is a neurodegenerative disease with devastating consequences for the patient and his/her family. Its etiology is still not clear. In about 10 % of the patients there is a hereditary pattern of the disease. Worldwide, prevalence ranges from 2 to 11 cases per 100,000 people. Age of presentation varies from 58 to 63 years for sporadic cases, and from 47 to 52 years for the familial ones. Concerning gender, there is a slight preference for males. Clinical manifestations include signs of upper and lower motor neurons, damage in limbs and bulbar muscles, and, in some patients, frontotemporal cognitive dysfunction. Diagnosis is essentially clinical supported by neurophysiological studies, such as needle electromyography, which is the most important test for early diagnosis. There is no cure, but riluzol has proven to delay the use of mechanical ventilation and to slightly prolong survival. Consequently, management is based on support measures, such as those related to nutrition and ventilatory function, in addition to control of the motor and non-motor symptoms of the disease.
KEY WORDS
Amyotrophic Lateral Sclerosis, Epidemiology, Motor Neuron Disease, Signs and Symptoms, Diagnosis, Therapeutics
RESUMO
La esclerose lateral amiotrófica é uma doença neurodegenerativa que tem consequências devastadoras para o doente e sua família. Ainda não existe claridade sobre sua etiologia, cerca de 10 % dos doentes tem padrão hereditário. A prevalência mundial varia entre 2 e 11 casos por 100.000 habitantes; a faixa de idade de apresentação é de 58 a 63 anos para os casos esporádicos, e de 47 a 52 anos para os familiares, com uma ligeira predileção pelo sexo masculino. As manifestações clínicas incluem signos de dano dos neurônios motores superior e inferior tanto nas extremidades como na musculatura bulbar, e em alguns doentes há deterioro cognitivo fronto-temporal. O diagnóstico continua sendo fundamentalmente clínico, apoiado por estudos neurofisiológicos; destes, a eletromiografia de agulha há sido o mais útil para o diagnóstico precoce. Não existe tratamento curativo e só um medicamento, o Riluzol, há demostrado efeito para retrasar o uso de ventilação mecânica e prolongar levemente a supervivência. Por tanto, o tratamento destes doentes se baseia em medidas de suporte, especialmente nos aspectos de nutrição e ventilação, ademais de controlar os sintomas motores e não motores da doença.
PALAVRAS CHAVE
Diagnóstico, Doença do Neurônio Motor, Epidemiologia, Esclerose Lateral Amiotrófica, Sinais e Sintomas, Terapêutica
Cómo citar: Zapata-Zapata CH, Franco-Dager E, Solano-Atehortúa JM, Ahunca-Velásquez LF. Esclerosis lateral amiotrófica: actualización. Iatreia. 2016 Abr-Jun;29(2):194-205. DOI 10.17533/udea.iatreia.v29n2a08.
INTRODUCCIÓN
La esclerosis lateral amiotrófica (ELA) es una enfermedad caracterizada por la degeneración progresiva de las neuronas motoras superior (NMS) e inferior (NMI), lo que produce debilidad de los músculos de las extremidades, torácicos, abdominales y bulbares (1). Entre 1865 y 1869 Jean Martin Charcot en sus estudios clínico-patológicos observó la correlación entre los signos clínicos piramidales y las lesiones de los cordones laterales, con la amiotrofia y las alteraciones en las astas anteriores de la médula espinal, por lo que en 1874 le dio el nombre de esclerosis lateral amiotrófica. Recientes avances científicos han permitido mejorar nuestro entendimiento sobre esta enfermedad, pero muchas dudas permanecen sin resolver. Este artículo resume los conceptos actuales sobre la etiología, los factores de riesgo, las manifestaciones clínicas, el diagnóstico, el tratamiento y el pronóstico.
ETIOPATOGENIA
Después de 140 años de la descripción inicial de la ELA, la etiopatogenia continúa sin aclararse completamente. Actualmente se la puede clasificar como familiar (ELAF) y esporádica (ELAS) (2). En ambos casos se produce un proceso neurodegenerativo que lleva a la muerte de las neuronas motoras. En la actualidad se considera que esta degeneración es un proceso focal de la NMS y la NMI que avanza continua y separadamente para sumarse en el tiempo (3). Excepto para algunos casos de ELAF, aún no se conoce la causa que desencadena el comienzo de los cambios fisiopatológicos e histopatológicos observados en esta enfermedad. El amplio espectro de posibles causas o consecuencias incluye, entre otras, las siguientes: estrés oxidativo (4), factores genéticos (2), excitotoxicidad por glutamato (5), daño mitocondrial (6), defecto en el transporte axonal (7), daño originado por los astrocitos (8) y apoptosis (9,10).
Factores ambientales
Se han investigado diversos factores ambientales que contribuyen al desarrollo de ELA (11). Se ha propuesto el tabaquismo como un posible factor de riesgo, pero la evidencia es débil. En un estudio de casos y controles en los Países Bajos entre el 2006 y el 2009, se encontró OR (Odds Ratio) de 1,38 (IC95 %: 1,02-1,88); además encontraron disminución en el tiempo de supervivencia en los pacientes con ELA que fumaban: HR (Hazard Ratio) de 1,51 (IC95 %: 1,07-2,15 (12). Sin embargo, dichas medidas de asociación fueron poco significativas.
En dos revisiones sistemáticas de la literatura se encontraron solo dos estudios que señalaban la exposición a pesticidas como factor de riesgo para desarrollar ELA, uno con OR de 2,5 (IC95 %: 1,2-5,1), y el otro, con OR de 1,23 (IC95 %: 1,03-1,46); los autores concluyeron que los pesticidas se identifican como un factor de riesgo para desarrollar ELA, pero que se necesitan estudios con mayor calidad metodológica para definir dicha relación (13).
En un estudio de Chío y colaboradores se observó que la actividad física de alto rendimiento (fútbol profesional) era un factor de riesgo para desarrollar ELA, independientemente de la edad (14). Sin embargo, en otro estudio del mismo autor no se encontraron casos de ELA en deportistas de alto rendimiento en otros deportes (15), lo cual parece indicar que la actividad física no es un factor de riesgo para desarrollar esta enfermedad y que se deben analizar otras variables en los jugadores de fútbol profesional.
Se han descrito otros factores ambientales asociados a ELA como el trauma craneoencefálico (16), la contaminación del agua en la isla Oshima (17) y la asociación espacial entre las zonas de residencia de pacientes con ELA y posibles factores de riesgo ambiental, como lo reportaron Caller y colaboradores al norte de Nueva Inglaterra, Estados Unidos (18). Sin embargo, en ninguno de estos se ha encontrado una relación causal directa.
Genética
Una mayor concordancia entre gemelos y la agregación familiar de trastornos neurodegenerativos son argumentos a favor de la existencia de factores genéticos de susceptibilidad para desarrollar ELA, pero dichos factores no se han podido definir claramente en la ELAS (2). Para el caso de la ELAF la mayoría de las mutaciones se transmiten de forma autosómica dominante (19); el primer gen que se relacionó con ELAF fue el de la superóxido-dismutasa 1, pero su alteración solo explica el 20 % de los casos de ELAF (20). La mutación del gen para la proteína TDP-43 se ha relacionado con el 10 % de los casos de ELAF y con demencia frontotemporal (DFT) (21). Se han descrito 13 mutaciones del gen FUS/TLP que en conjunto explican aproximadamente el 5 % de los casos de ELAF (22). Recientemente se ha correlacionado la sobreexpresión del hexanucleótido GGGGCC en el gen ubicado en el cromosoma 9 (C9ORF72), con mayor predisposición a encontrar en un mismo individuo ELAF y DFT (23). En la actualidad se considera esta sobreexpresión como la principal causa genética de ELAF (24) y recientemente se le atribuye un papel relevante en casos de ELAS (25).
EPIDEMIOLOGÍA
En la actualidad no existe una prueba o biomarcador definitorio de ELA, el diagnóstico continúa siendo principalmente clínico y no existe un período prodrómico claro, lo cual dificulta los estudios epidemiológicos (26). Sin embargo, el aumento en el conocimiento de la enfermedad y el uso de criterios estandarizados para el diagnóstico han incrementado el registro de los casos. En una revisión sistemática de la literatura se observó una gran variabilidad en las cifras de prevalencia, que van desde 2,0/100.000 habitantes en China hasta 11,3/100.000 habitantes en Japón, con cifras intermedias para Europa y Norteamérica. Similar variabilidad encontraron en las cifras de incidencia (tabla 1). Los autores señalan que la gran variabilidad en los datos se puede deber a diferencias en el diseño metodológico de los estudios, pero no descartan que se deban a diferencias poblacionales como la edad, los factores ambientales y la predisposición genética (27).
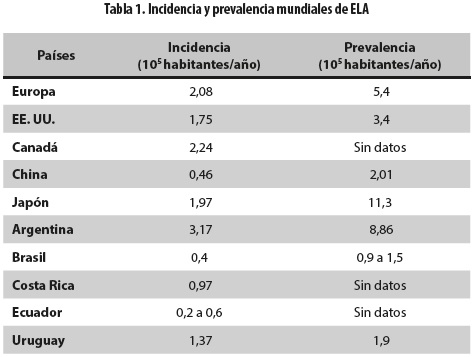
En un estudio de países europeos se encontró mayor incidencia en hombres, con dos picos de edad de presentación: 58 a 63 años para los casos esporádicos y 47 a 52 años para los familiares; la enfermedad fue muy rara después de los 80 años (28). Cronin y colaboradores hicieron una revisión sistemática de la literatura (1966 a 2006), acerca de la variación étnica en ELA y encontraron que la incidencia es más baja en etnias africanas, asiáticas e hispánicas con respecto a caucásicos europeos y norteamericanos (29).
En América Latina se han hecho pocos estudios sobre ELA (tabla 1); se dispone de algunos datos epidemiológicos de incidencia (casos/100.000 habitantes/año) y prevalencia (casos/100.000 habitantes), respectivamente, así: Argentina 3,17 y 8,86 (30); Brasil 0,4 y 0,9 a 1,5 (31); Costa Rica 0,97 y sin datos (32); Ecuador 0,2 a 0,6 y sin datos (33) y Uruguay 1,37 y 1,9 (34). No se encontraron publicaciones en México, Cuba y Colombia sobre ninguno de los dos datos mencionados (35-37).
MANIFESTACIONES CLÍNICAS
La forma clásica de esta enfermedad se caracteriza por la mezcla de manifestaciones clínicas de lesión de las neuronas motoras superior e inferior y signos de alteración bulbar y respiratoria (tabla 2) (38).
Las siguientes claves clínicas apoyan el diagnóstico de ELA: comienzo lento, poco llamativo y asimétrico, alteración de varios segmentos corporales, déficit de la NMS o la NMI y síntomas o signos de disfunción bulbar. Es frecuente que existan signos de liberación piramidal en sitios por encima de aquellos en donde hay atrofia muscular (39). Muchos pacientes con ELA presentan síntomas de déficit cognitivo, conductual y comportamental en el espectro de la DFT; estos son principalmente disfunción ejecutiva, irritabilidad, cambios de la personalidad con impulsividad y mal reconocimiento de la enfermedad; su presencia constituye un marcador pronóstico negativo. La apatía, la desinhibición y el mal control social también hacen parte de este espectro (40). En un estudio comparativo del perfil cognitivo y conductual de los pacientes con ELA confirmada se halló lo siguiente: 48 % no tenían cambios cognitivo-conductuales, 31 % tenían déficit cognitivo puro, 9 % cumplían los criterios de la DFT, 4 % tenían alteración conductual pura y 4 % tenían diagnóstico previo de enfermedad de Alzheimer (41).
Patrones clínicos
ELA clásica: representa el 65 % a 70 % de los casos; el pico de edad de comienzo es de 58 a 63 años. En este patrón hay afectación de las neuronas motoras superior e inferior y, en un principio, de las extremidades, con diseminación posterior al resto de la musculatura corporal incluyendo alteración bulbar y por último falla respiratoria (26).
Esclerosis lateral primaria (ELP): representa el 20 % de los casos de las enfermedades motoneuronales en el adulto; se debe al daño de la NMS sin ningún signo de la NMI. Comienza con paraparesia espástica pura y con el transcurso del tiempo va afectando los brazos, las manos y los músculos orofaríngeos. El 50 % de los pacientes pueden tener espasticidad de la vejiga urinaria. Pringle y colaboradores (42) sugieren que un criterio diagnóstico de esta variante sea el progreso de la enfermedad durante tres años sin signos de NMI; sin embargo, muchos pacientes con diagnóstico de ELP desarrollan signos de lesión de la NMI después de muchos años de haber comenzado la enfermedad, por lo que algunos expertos prefieren denominar este trastorno como ''ELA con predominio de daño de NMS''. La progresión es mucho más lenta que en los casos de ELA clásica y la supervivencia es mayor (43).
Atrofia muscular progresiva (AMP): en esta variante de ELA hay únicamente signos de lesión de la NMI. Es más común en varones que en mujeres (relación 4:1). La velocidad de progresión es muy variable, por lo general más lenta que en la forma clásica. Se han documentado casos de supervivencia de 15 años o más. Contrario a lo que sucede en la ELP, muchos pacientes con AMP pueden presentar signos de daño de la NMS después de muchos años de haber comenzado la enfermedad, por lo que se la ha denominado ''ELA con predominio de daño de la NMI'' (44).
Parálisis bulbar progresiva (PBP): este patrón constituye del 25 % al 30 % de los casos de ELA. Se caracteriza por comienzo y predominio del síndrome bulbar, con o sin signos de liberación piramidal. Generalmente hay disartria y disfagia, con atrofia y fasciculaciones linguales. También pueden aparecer tempranamente debilidad espástica del maxilar inferior y cierre involuntario de la mandíbula por el espasmo; el tiempo de evolución y supervivencia es de uno a dos años. La mayoría de los pacientes con PBP llegan a desarrollar la forma clásica de ELA (45).
ESTUDIOS DIAGNÓSTICOS
A todos los pacientes se les deben hacer los siguientes estudios diagnósticos:
Neurofisiología: la electromiografía con neuroconducciones ha sido la ayuda más útil para el diagnóstico y seguimiento de pacientes con enfermedades de las neuronas motoras y para su diagnóstico diferencial (46); por medio de ella se logra detectar alteraciones subclínicas de la NMI.
Laboratorio: los estudios de laboratorio clínico ayudan a descartar otros trastornos que pueden simular síndromes de las neuronas motoras, comorbilidades y complicaciones de la enfermedad. Se recomiendan los siguientes: hemograma, reactantes de fase aguda, pruebas de función renal, hepática y tiroidea, electrólitos, electroforesis de proteínas y perfil glucémico. Además, en casos seleccionados, estudios del líquido cefalorraquídeo, de histopatología, biología molecular, genética y otros (47).
Neuroimágenes: el principal papel de las neuroimágenes en pacientes con ELA es descartar otras causas de un síndrome piramidal como tumores del neuroeje, radiculopatías, enfermedad cerebrovascular, mielopatías, etc. No existe un patrón imaginológico específico para la ELA y en estos pacientes las neuroimágenes son, generalmente, normales. Sin embargo, a medida que avanza la enfermedad, la resonancia magnética (RM) puede mostrar atrofia cortical de predominio frontotemporal y en el segmento anterior de la médula espinal. En la RM convencional se ha descrito clásicamente hiperintensidad del tracto corticoespinal en las secuencias T2 y FLAIR, al igual que hipointensidad en la corteza precentral (48). Aún no se ha definido la utilidad clínica de otros estudios como la tractografía, la fracción de anisotropía, la espectroscopia y la difusibilidad media (49).
Estudios genéticos: las pruebas genéticas no se solicitan de rutina en pacientes con ELA, pero se debe solicitar la asesoría genética en casos de ELAF o de enfermedad de inicio juvenil (50).
CRITERIOS DIAGNÓSTICOS
La evolución de los criterios diagnósticos ha permitido aumentar el número de pacientes a los que se les diagnostica esta enfermedad. En 1994 se crearon los primeros criterios aceptados para ELA, llamados Criterios de El Escorial (51), se revisaron en 1997, Criterios de El Escorial Revisados (CEER) (52). El diagnóstico de ELA se basa en su aplicación, para determinar la presencia de enfermedad de la NMS evidenciada clínicamente y de la NMI demostrada clínica o electromiográficamente; los pacientes se clasifican de acuerdo con el número de regiones corporales afectadas de un total de cuatro: bulbar, cervical, torácica y lumbar. Estos criterios tienen baja sensibilidad, e incluso se sabe que muchos pacientes mueren por esta enfermedad sin llegar a cumplirlos (53); por esta razón, los CEER fueron modificados en el 2008 con el algoritmo de Awaji-Shima (figura 1) (54), en el que se hicieron algunos cambios, pero se mantuvieron los principios de los criterios CEER. En dicho algoritmo se clasificó la certeza diagnóstica en tres categorías: clínicamente posible, clínicamente probable y clínicamente definida; los criterios de Awaji-Shima tienen mayor sensibilidad (81 % versus 62 %), e igual especificidad (95 %) que los CEER (46).
TRATAMIENTO
No existe tratamiento curativo para la ELA. Se han ensayado muchas sustancias sin demostrar efectividad (55). La investigación actual se está enfocando en la manipulación de algunas proteínas musculares, los factores de crecimiento neuronal, la terapia de reemplazo celular y la terapia génica que busca el silenciamiento de genes (56).
Actualmente el principal objetivo del tratamiento es prolongar la supervivencia y mejorar la calidad de vida de los pacientes. Así, el mejor tratamiento es una combinación de agentes neuroprotectores, manejo sintomático, nutricional y soporte ventilatorio. Existe evidencia de que el tratamiento por un equipo multidisciplinario experto en el manejo de estos pacientes mejora la calidad de vida y prolonga la supervivencia (57).
El equipo es coordinado por el neurólogo especialista en esta área y debe contar con enfermera, terapeuta físico y ocupacional, nutricionista, fonoaudiólogo, psicólogo y trabajador social; en ocasiones se requieren también neumólogo, psiquiatra y ortopedista.
El único medicamento aprobado por la FDA (Food and Drug Administration) y el INVIMA (Instituto Nacional de Vigilancia de Alimentos y Medicamentos de Colombia) es el riluzol, un antagonista de los receptores de N-metil D-aspartato (NMDA) y se supone que reduce la excitotoxicidad en la ELA (58). En un ensayo clínico con asignación aleatoria se demostró que una dosis de 100 mg al día prolongó la vida 18 meses (59). Los pacientes que más se benefician de este medicamento son los que han tenido un curso menor de cinco años, con diagnóstico probable o definitivo, y sin traqueostomía. Este medicamento no ha demostrado beneficio en mejorar la función motora, las fasciculaciones, ni la función ventilatoria (60).
Tratamiento sintomático: el objetivo del tratamiento sintomático es mejorar la calidad de vida de los pacientes y cuidadores. Se debe tener en cuenta una gran cantidad de síntomas en el manejo de los pacientes con ELA; en el caso de la sialorrea, son de utilidad los antidepresivos tricíclicos, las gotas de atropina y, en caso de refractariedad, la toxina botulínica (61). En el paciente con afectación pseudobulbar, el dextrometorfano combinado con quinidina (62); los antidepresivos tricíclicos o los inhibidores selectivos de la recaptación de serotonina han tenido efectividad en el control de los síntomas emocionales (63). La espasticidad se puede controlar con terapias físicas (64) y si es persistentes o grave, es apropiado usar relajantes musculares como baclofeno y tizanidina (65).
Soporte ventilatorio: la insuficiencia respiratoria es la principal causa de muerte de los pacientes con ELA. Para ella, la ventilación no invasiva (VNI) es una medida terapéutica efectiva; el método más fisiológico es la ventilación con presión positiva intermitente binivel (BiPAP). Las guías internacionales recomiendan su prescripción ante la presencia de síntomas relacionados con la insuficiencia respiratoria asociada a uno de los siguientes hallazgos (59): PaCO2 mayor de 45 mm Hg, capacidad vital menor de 50 % de la normal, presión inspiratoria máxima por debajo de 60 % de la normal, desaturación nocturna de PaO2 por debajo del 90 % más de 5 % del tiempo. La VNI y la aspiración de secreciones mejoran la calidad del sueño y la función cognitiva, prolongan la supervivencia y mejoran la calidad de vida (60).
Nutrición: a medida que avanza la enfermedad, muchos factores llevan a un balance negativo de calorías, de lo que resulta un deterioro nutricional variable; el índice de pérdida de peso puede ser un predictor de la velocidad de progresión (66). En caso de dificultad para deglutir la dieta mixta, se debe pasar a una dieta supraglótica con posturas adecuadas para facilitar la deglución. Cuando han fracasado las medidas anteriores es necesario hacer gastrostomía percutánea; con esta última se ha demostrado mejoría del estado nutricional y la calidad de vida, pero sin prolongar la supervivencia (67).
Factores de pronóstico: en una revisión sistemática de la literatura se encontraron los siguientes factores de mal pronóstico: edad avanzada (por encima de 80 años), forma de presentación bulbar, período corto de latencia, progresión rápida de la enfermedad, factores sicosociales relacionados con el ánimo y la calidad de vida, alteraciones de la función cognitiva, mal estado nutricional (índice de masa corporal menor de 18,5) y baja capacidad vital forzada (menor del 50 %). El tiempo promedio de supervivencia de pacientes con ELA está entre 3 y 5 años desde el inicio de los síntomas (68).
CONCLUSIONES
La ELA es una enfermedad neurodegenerativa caracterizada por daño progresivo de las neuronas motoras. La mayor parte de los casos son esporádicos, aunque ha venido en aumento el descubrimiento de casos con patrón de transmisión familiar. La etiopatogenia continúa sin aclararse, y solo existen piezas del rompecabezas en construcción. Los estudios epidemiológicos mundiales son muy pocos y Colombia no se aleja de dicha situación. La presentación clínica es heterogénea, y el diagnóstico continúa siendo clínico; sin embargo, las ayudas diagnósticas sirven para disminuir el tiempo de latencia y descartar causas secundarias. El tratamiento debe ser multidisciplinario ofreciendo a todos los pacientes la oportunidad de recibir el riluzol, que parece prolongar la supervivencia, además de dar soporte nutricional y ventilatorio cuando sea necesario. Los avances en genética y en el conocimiento de la fisiopatología de la enfermedad están abriendo nuevas posibilidades terapéuticas; entre ellas se destacan el reemplazo celular, la terapia génica y la prevención del acúmulo de agregados proteicos. Hasta entonces, los pacientes deben recibir la mejor atención, con el objetivo de mantener la mayor comodidad y dignidad posibles en el trascurso de esta catastrófica enfermedad.
CONFLICTOS DE INTERÉS
Ninguno que declarar.
AGRADECIMIENTOS
A Jenny García Valencia, Profesora Asociada del Departamento de Psiquiatría, Facultad de Medicina. Universidad de Antioquia, por su apoyo en la revisión de la literatura.
REFERENCIAS BIBLIOGRÁFICAS
1. Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011 Oct;7(11):639-49. DOI 10.1038/nrneurol.2011.153. [ Links ]
2. Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011 Oct;7(11):603-15. DOI 10.1038/nrneurol.2011.150. [ Links ]
3. Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009 Sep;73(10):805-11. DOI 10.1212/WNL.0b013e3181b6bbbd. [ Links ]
4. D'Amico E, Factor-Litvak P, Santella RM, Mitsumoto H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic Biol Med. 2013 Dec;65:509-27. DOI 10.1016/j.freeradbiomed.2013.06.029. [ Links ]
5. Spreux-Varoquaux O, Bensimon G, Lacomblez L, Salachas F, Pradat PF, Le Forestier N, et al. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci. 2002 Jan;193(2):73-8. [ Links ]
6. Menzies FM, Ince PG, Shaw PJ. Mitochondrial involvement in amyotrophic lateral sclerosis. Neurochem Int. 2002 May;40(6):543-51. [ Links ]
7. Tomkins J, Usher P, Slade JY, Ince PG, Curtis A, Bushby K, et al. Novel insertion in the KSP region of the neurofilament heavy gene in amyotrophic lateral sclerosis (ALS). Neuroreport. 1998 Dec;9(17):3967-70. [ Links ]
8. Julien JP. ALS: astrocytes move in as deadly neighbors. Nat Neurosci. 2007 May;10(5):535-7. [ Links ]
9. Sathasivam S, Ince PG, Shaw PJ. Apoptosis in amyotrophic lateral sclerosis: a review of the evidence. Neuropathol Appl Neurobiol. 2001 Aug;27(4):257-74. [ Links ]
10. Shaw PJ. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry. 2005 Aug;76(8):1046-57. [ Links ]
11. Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, et al. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav. 2013 Jul;3(4):431-57. DOI 10.1002/brb3.142. [ Links ]
12. de Jong SW, Huisman MH, Sutedja NA, van der Kooi AJ, de Visser M, Schelhaas HJ, et al. Smoking, alcohol consumption, and the risk of amyotrophic lateral sclerosis: a population-based study. Am J Epidemiol. 2012 Aug;176(3):233-9. DOI 10.1093/aje/kws015. [ Links ]
13. Sutedja NA, Veldink JH, Fischer K, Kromhout H, Heederik D, Huisman MH, et al. Exposure to chemicals and metals and risk of amyotrophic lateral sclerosis: a systematic review. Amyotroph Lateral Scler. 2009 Oct- Dec;10(5-6):302-9. DOI 10.3109/17482960802455416. [ Links ]
14. Chiò A, Benzi G, Dossena M, Mutani R, Mora G. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 2005 Mar;128(Pt 3):472-6. [ Links ]
15. Chiò A, Calvo A, Dossena M, Ghiglione P, Mutani R, Mora G. ALS in Italian professional soccer players: the risk is still present and could be soccer-specific. Amyotroph Lateral Scler. 2009 Aug;10(4):205-9. DOI 10.1080/17482960902721634. [ Links ]
16. Armon C, Nelson LM. Is head trauma a risk factor for amyotrophic lateral sclerosis? An evidence based review. Amyotroph Lateral Scler. 2012 Jun;13(4):351-6. DOI 10.3109/17482968.2012.660954. [ Links ]
17. Kihira T, Okamoto K, Yoshida S, Kondo T, Iwai K, Wada S, et al. Environmental characteristics and oxidative stress of inhabitants and patients with amyotrophic lateral sclerosis in a high-incidence area on the Kii Peninsula, Japan. Intern Med. 2013;52(13):1479-86. [ Links ]
18. Caller TA, Chipman JW, Field NC, Stommel EW. Spatial analysis of amyotrophic lateral sclerosis in Northern New England, USA, 1997-2009. Muscle Nerve. 2013 Aug;48(2):235-41. DOI 10.1002/mus.23761. [ Links ]
19. Siddique T, Figlewicz DA, Pericak-Vance MA, Haines JL, Rouleau G, Jeffers AJ, et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med. 1991 May;324(20):1381-4. [ Links ] Erratum in: N Engl J Med. 1991 Jul;325(1):71.
20. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993 Mar;362(6415):59- 62. [ Links ] Erratum in: Nature. 1993 Jul;364(6435):362.
21. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006 Dec;351(3):602-11. [ Links ]
22. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009 Feb;323(5918):1205-8. DOI 10.1126/science.1166066. [ Links ]
23. De Jesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011 Oct;72(2):245-56. DOI 10.1016/j.neuron.2011.09.011. [ Links ]
24. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011 Oct;72(2):257-68. DOI 10.1016/j.neuron.2011.09.010. [ Links ]
25. Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010 Oct;9(10):986-94. DOI 10.1016/S1474-4422(10)70197-6. [ Links ] Erratum in: Lancet Neurol. 2011 Mar;10(3):205.
26. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. 2011 Mar;377(9769):942-55. [ Links ] DOI 10.1016/S0140-6736(10)61156-7.
27. Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118-30. DOI 10.1159/000351153. [ Links ]
28. Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010 Apr;81(4):385-90. DOI 10.1136/jnnp.2009.183525. [ Links ]
29. Cronin S, Hardiman O, Traynor BJ. Ethnic variation in the incidence of ALS: a systematic review. Neurology. 2007 Mar;68(13):1002-7. [ Links ]
30. Bettini M, Vicens J, Giunta DH, Rugiero M, Cristiano E. Incidence and prevalence of amyotrophic lateral sclerosis in an HMO of Buenos Aires, Argentina. Amyotroph Lateral Scler Frontotemporal Degener. 2013 Dec;14(7-8):598-603. DOI 10.3109/21678421.2013.808225. [ Links ]
31. Dietrich-Neto F, Callegaro D, Dias-Tosta E, Silva HA, Ferraz ME, Lima JM, et al. Amyotrophic lateral sclerosis in Brazil: 1998 national survey. Arq Neuropsiquiatr. 2000 Sep;58(3A):607-15. [ Links ]
32. Rodríguez-Paniagua P, Salas-Herrera I, Cartín-Brenes M. Incidencia de esclerosis lateral amiotrófica en Costa Rica. Acta Méd Costarric. 2007 Ene-Mar;49(1):33-7. [ Links ]
33. Bucheli M, Andino A, Montalvo M, Cruz J, Atassi N, Berry J, et al. Amyotrophic lateral sclerosis: analysis of ALS cases in a predominantly admixed population of Ecuador. Amyotroph Lateral Scler Frontotemporal Degener. 2014 Mar;15(1-2):106-13. DOI 10.3109/21678421.2013.852590. [ Links ]
34. Vázquez MC, Ketzoián C, Legnani C, Rega I, Sánchez N, Perna A, et al. Incidence and prevalence of amyotrophic lateral sclerosis in Uruguay: a populationbased study. Neuroepidemiology. 2008;30(2):105-11. [ Links ] DOI 10.1159/000120023.
35. Martínez HR, Molina-López JF, Cantú-Martínez L, González-Garza MT, Moreno-Cuevas JE, Couret- Alcaraz P, et al. Survival and clinical features in Hispanic amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler. 2011 May;12(3):199-205. DOI 10.3109/17482968.2010.550302. [ Links ]
36. Zaldivar T, Gutierrez J, Lara G, Carbonara M, Logroscino G, Hardiman O. Reduced frequency of ALS in an ethnically mixed population: a population-based mortality study. Neurology. 2009 May;72(19):1640-5. DOI 10.1212/WNL.0b013e3181a55f7b. [ Links ]
37. Ortiz Corredor F, Mendoza Pulido C, Peña Preciado M. Diseño de un sistema de clasificación para evaluar el grado de discapacidad de los pacientes con esclerosis lateral amiotrófica. Rev Col Med Fis Rehab. 2011;21(1):14-22. [ Links ]
38. Riku Y, Atsuta N, Yoshida M, Tatsumi S, Iwasaki Y, Mimuro M, et al. Differential motor neuron involvement in progressive muscular atrophy: a comparative study with amyotrophic lateral sclerosis. BMJ Open. 2014 May;4(5):e005213. DOI 10.1136/bmjopen-2014-005213. [ Links ]
39. Chieia MA, Oliveira AS, Silva HC, Gabbai AA. Amyotrophic lateral sclerosis: considerations on diagnostic criteria. Arq Neuropsiquiatr. 2010 Dec;68(6):837-42. [ Links ]
40. Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009 Jun;10(3):131-46. [ Links ] Erratum in: Amyotroph Lateral Scler. 2009 Aug;10(4):252.
41. Consonni M, Iannaccone S, Cerami C, Frasson P, Lacerenza M, Lunetta C, et al. The cognitive and behavioural profile of amyotrophic lateral sclerosis: application of the consensus criteria. Behav Neurol. 2013;27(2):143-53. DOI 10.3233/BEN-2012-110202. [ Links ]
42. Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. 1992 Apr;115 ( Pt 2):495-520. [ Links ]
43. D'Amico E, Pasmantier M, Lee YW, Weimer L, Mitsumoto H. Clinical evolution of pure upper motor neuron disease/dysfunction (PUMMD). Muscle Nerve. 2013 Jan;47(1):28-32. DOI 10.1002/mus.23496. Erratum in: Muscle Nerve. 2014 Oct;50(4):619. [ Links ]
44. Kim WK, Liu X, Sandner J, Pasmantier M, Andrews J, Rowland LP, et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. 2009 Nov;73(20):1686-92. DOI 10.1212/WNL.0b013e3181c1dea3. [ Links ]
45. Karam C, Scelsa SN, Macgowan DJ. The clinical course of progressive bulbar palsy. Amyotroph Lateral Scler. 2010 Aug;11(4):364-8. DOI 10.3109/17482960903513159. [ Links ]
46. Costa J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis:a systematic review. Arch Neurol. 2012 Nov;69(11):1410-6. [ Links ]
47. EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis; Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol. 2012 Mar;19(3):360-75. DOI 10.1111/j.1468-1331.2011.03501.x. [ Links ]
48. Alvarez-Uría Tejero MJ, Sáiz Ayala A, Fernández Rey C, Santamarta Liébana ME, Costilla García S. Diagnóstico de la esclerosis lateral amiotrófica: avances en RM. Radiologia. 2011 Mar-Apr;53(2):146-55. DOI 10.1016/j.rx.2010.10.004. [ Links ]
49. Wang S, Poptani H, Bilello M, Wu X, Woo JH, Elman LB, et al. Diffusion tensor imaging in amyotrophic lateral sclerosis: volumetric analysis of the corticospinal tract. AJNR Am J Neuroradiol. 2006 Jun-Jul;27(6):1234-8. [ Links ]
50. Chiò A, Battistini S, Calvo A, Caponnetto C, Conforti FL, Corbo M, et al. Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry. 2014 May;85(5):478-85. DOI 10.1136/jnnp-2013-305546. [ Links ]
51. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/ Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial ''Clinical limits of amyotrophic lateral sclerosis'' workshop contributors. J Neurol Sci. 1994 Jul;124 Suppl:96-107. [ Links ]
52. Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000 Dec;1(5):293-9. [ Links ]
53. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman OM. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: A population-based study. Arch Neurol. 2000 Aug;57(8):1171-6. [ Links ]
54. de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008 Mar;119(3):497-503. DOI 10.1016/j.clinph.2007.09.143. [ Links ]
55. Aggarwal S, Cudkowicz M. ALS drug development: reflections from the past and a way forward. Neurotherapeutics. 2008 Oct;5(4):516-27. DOI 10.1016/j.nurt.2008.08.002. [ Links ]
56. Gordon PH. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013 Oct;4(5):295-310. DOI 10.14336/AD.2013.0400295. [ Links ]
57. Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000. J Neurol Neurosurg Psychiatry. 2003 Sep;74(9):1258-61. [ Links ]
58. Couratier P, Sindou P, Esclaire F, Louvel E, Hugon J. Neuroprotective effects of riluzole in ALS CSF toxicity. Neuroreport. 1994 Apr;5(8):1012-4. [ Links ]
59. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994 Mar;330(9):585-91. [ Links ]
60. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012 Mar;3:CD001447. DOI 10.1002/14651858.CD001447. [ Links ]
61. Giess R, Naumann M, Werner E, Riemann R, Beck M, Puls I, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhoea in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2000 Jul;69(1):121-3. [ Links ]
62. Pioro EP, Brooks BR, Cummings J, Schiffer R, Thisted RA, Wynn D, et al. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010 Nov;68(5):693-702. DOI 10.1002/ana.22093. [ Links ]
63. Szczudlik A, Slowik A, Tomik B. [The effect of amitriptyline on the pathological crying and other pseudobulbar signs]. Neurol Neurochir Pol. 1995 Sep-Oct;29(5):663-74. [ Links ] Polish.
64. Drory VE, Goltsman E, Reznik JG, Mosek A, Korczyn AD. The value of muscle exercise in patients with amyotrophic lateral sclerosis. J Neurol Sci. 2001 Oct;191(1-2):133-7. [ Links ]
65. McClelland S 3rd, Bethoux FA, Boulis NM, Sutliff MH, Stough DK, Schwetz KM, et al. Intrathecal baclofen for spasticity-related pain in amyotrophic lateral sclerosis: efficacy and factors associated with pain relief. Muscle Nerve. 2008 Mar;37(3):396-8. [ Links ]
66. Marin B, Desport JC, Kajeu P, Jesus P, Nicolaud B, Nicol M, et al. Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. 2011 Jun;82(6):628-34. DOI 10.1136/jnnp.2010.211474. [ Links ]
67. Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidencebased review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009 Oct;73(15):1227-33. DOI 10.1212/WNL.0b013e3181bc01a4. [ Links ]
68. Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009 Oct-Dec;10(5-6):310-23. DOI 10.3109/17482960802566824. [ Links ]

