










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.29 no.2 Medellín Apr./June 2016
https://doi.org/10.17533/udea.iatreia.v29n2a10
DOI 10.17533/udea.iatreia.v29n2a10
PRESENTACIÓN DE CASO
Estudio de la braquidactilia en población gitana: descripción de un caso familiar
Study of brachydactyly in gipsy population: Description of a familial case
Estudo da braquidactilia da população cigana: Descrição de um caso familiar
Antonia Pérez-Lázaro1; Víctor Alché-Ramírez2; Ana María Núñez-Negrillo3; Juan Francisco Gamella-Mora4; Rafael Fernández-Castillo5
1 Enfermera, Distrito Sanitario Guadix, Servicio Andaluz de Salud, Granada, España.
2 Médico Especialista, Distrito Sanitario Guadix, Servicio Andaluz de Salud, Granada, España.
3 Profesora, Facultad de Ciencias de la Salud, Departamento de Enfermería, Universidad de Granada, España.
4 Doctor, Departamento de Antropología, Facultad de Filosofía y Letras, Universidad de Granada, España.
5 Doctor, Departamento de Enfermería, Facultad de Ciencias de la Salud, Universidad de Granada, España. rafaelfernandez@ugr.es
Recibido: abril 10 de 2015
Aceptado: julio 28 de 2015
RESUMEN
La braquidactilia constituye una malformación genética heredable con carácter autosómico dominante o recesivo. En este artículo se describe el caso de una familia gitana que presenta braquidactlia congénita. El estudio se hizo en el Distrito Sanitario de Guadix en Granada. Los sujetos de estudio fueron cuatro hermanos (dos hombres y dos mujeres) integrantes de la misma unidad familiar y pertenecientes a la comunidad gitana. Se recogieron datos sociodemográficos y genéticos. Los sujetos presentan la manifestación de braquidactilia expresada fenotípicamente con alguna variabilidad entre ellos. Los datos radiológicos evidencian que corresponden a la braquidactilia tipo A4. Uno de ellos presenta una mezcla de A4 con E, o quizás se trate de una nueva variedad no clasificada. Todos presentan anomalías similares en los pies. Además, presentan obesidad, dislipidemia y diversos grados de consanguinidad.
PALABRAS CLAVE
Agentes Comunitarios de Salud, Braquidactilia, Consanguinidad, Grupos Étnicos, Salud Comunitaria
SUMMARY
Brachydactyly is an inheritable autosomal genetic malformation, either dominant or recessive. This article describes a gypsy family presenting with congenital brachydactyly. The study was conducted at the Sanitary District of Guadix, in Granada, Spain. The study subjects were four siblings (two women and two men), members of the same family and belonging to the Roma community. Demographic and genetic data were collected. With some variability, they had the phenotypic manifestation of brachydactyly. Radiographic data revealed that it was type A4 brachydactyly, but one of them featured a blend of A4 with E, or perhaps it is a new unclassified variety. All cases showed similar abnormalities in the feet. Besides, they are obese, and have dyslipidemia and different degrees of consanguinity.
KEY WORDS
Brachydactyly, Community Health, Community Health Agents, Consanguinity, Ethnic Groups
RESUMO
A braquidactilia constitui uma malformação genética com caráter autossômico dominante ou recessiva. Este artigo descreve o caso de uma família cigana que apresenta braquidactlia congênitas. O estudo foi feito no Distrito de Sanitário de Guadix em Granada. Os sujeitos do estudo foram quatro irmãos (dois homens e duas mulheres) membros da mesma unidade familiar e pertencentes à comunidade cigana. Foram coletados dados demográficos e genéticos. Os sujeitos apresentam a manifestação de braquidactilia expressa fenotipicamente com alguma variabilidade entre eles. Os elementos radiológicos mostram que correspondem à braquidactilia tipo A4. Um deles apresenta uma mistura de A4, com E, ou, talvez, uma nova variedade não classificadas. Todos têm anomalias semelhantes nos pés. Ademais, apresentam obesidade, dislipidemia e diferentes graus de consanguinidade.
PALAVRAS CHAVE
Agentes Comunitários de Saúde, Braquidactilia, Consanguinidade, Grupos Étnicos, Saúde Comunitária
Cómo citar: Pérez-Lázaro A, Alché-Ramírez V, Núñez-Negrillo AM, Gamella-Mora JF, Fernández-Castillo R. Estudio de la braquidactilia en población gitana: descripción de un caso familiar. Iatreia. 2016 Abr-Jun;29(2):218-227. DOI 10.17533/udea.iatreia.v29n2a10.
INTRODUCCIÓN
A diferencia de otras poblaciones, cuya historia, genealogía y epidemiología genética han sido ampliamente documentadas, los gitanos han sido ignorados por la medicina europea debido a su dispersión geográfica y su marginalidad social. No obstante, su patrimonio genético único se ha convertido ahora en foco de interés para genetistas y médicos (1). La investigación genética médica ha revelado una peculiar combinación de homogeneidad genética, mutaciones compartidas y, al mismo tiempo, un mosaico de notables diferencias en la prevalencia de trastornos genéticos y mutaciones entre comunidades genéticas vecinas de un mismo país (2). Este patrón epidemiológico sugiere una estructura poblacional compleja, cuya comprensión es esencial para su posterior investigación genética, así como para la atención de los pacientes y las intervenciones de salud pública (3,4).
El papel de la atención primaria respecto a la genética será cada vez más importante, como lo demuestra el hecho de que clásicamente su función se ha limitado a informar sobre la etiología y patogenia de algunas enfermedades, pero que en la actualidad desempeña un papel activo, mediante la derivación a unidades especializadas o de genética, donde se hacen hoy en día el diagnóstico, la prevención y el tratamiento de muchas enfermedades (5,6). La población gitana presenta unos patrones reproductivos endogámicos y consanguíneos particulares; no obstante, se acepta igualmente que otros factores como los ambientales son fundamentales en el desarrollo de enfermedades ya sean genéticas o adquiridas (7). No existen evidencias claras de si determinadas afecciones genéticas inciden de igual manera en los distintos grupos sociales, si se relacionan también con otras variables o si, por el contrario, se pueden atribuir a desigualdades en salud (8). La braquidactilia constituye una malformación genética heredable con carácter autosómico dominante o recesivo, según el tipo concreto de que se trate. El gen responsable de esta anomalía está descrito solo para una minoría de las braquidactilias identificadas (9). Se caracteriza por la afectación de las falanges de los dedos de manos y pies (metacarpos/ tarsos), que puede oscilar desde un ligero acortamiento hasta su total ausencia. Puede ser perceptible a partir de las seis a ocho semanas de vida intrauterina con los medios técnicos disponibles actualmente que permiten el seguimiento del desarrollo fetal (10).
En general, el término braquidactilia incluye un grupo de enfermedades relacionadas. La literatura consultada evidencia que esta anomalía puede presentarse de forma aislada, es decir, afectando a un solo hueso de la mano, o bien, como cuadro sindrómico (11) con distintos niveles de expresividad morfológica y/o funcional manifestados a través del fenotipo de los individuos con diferentes niveles de variabilidad y morbimortalidad (12). Se ha catalogado la braquidactilia entre las enfermedades raras, y se han descrito diversos tipos de la misma en función de las extremidades afectadas. En general, todos los tipos de braquidactilia son de aparición infrecuente, a excepción del A3 y el D (13). A continuación se describen los diferentes tipos:
Braquidactilia tipo A: el acortamiento se limita a falanges medias. Dependiendo de los dedos afectados se forman diferentes subcategorías:
A1: conocida también como síndrome Farabee.
A2: conocida como síndrome Mohr-Wriedt.
A3: manifestada por el acortamiento de la falange media del quinto dedo. Se observa en el síndrome de Down.
A4: se considera un subtipo sumamente raro que suele afectar a las falanges medias del segundo y quinto dedos de las manos, es bilateral y puede expresarse también en los dedos de los pies, y acompañarse de ausencia de las falanges medias de los cuatro dedos laterales de ambos pies. Se ha descrito este subtipo como una entidad benigna, asintomática y no suele acarrear ningún tipo de discapacidad.
Quedan pendientes otras braquidactilias aún en fase de clasificación y que corresponderían posiblemente a las subcategorías A5, A6, A7.
Braquidactilia tipo B: suele manifestarse por el acortamiento de las falanges medias y terminales del segundo al quinto dedos. Puede haber sindactilia y ausencia de uñas.
Braquidactilia tipo C: se evidencia por el acortamiento de las falanges medias y proximales del segundo y tercer dedos.
Braquidactilia tipo D: es la de más frecuente aparición. Las falanges terminales de los pulgares son anchas y cortas.
Braquidactilia tipo E: asociada a retraso mental y otras malformaciones.
El objetivo de este trabajo fue describir, desde la atención primaria de salud, la situación sociosanitaria de una familia gitana que presentaba braquidactilia congénita. Este trabajo se hizo en el contexto de la prestación de asistencia sanitaria en atención primaria del Distrito Sanitario de Guadix en Granada. Los sujetos de estudio fueron cuatro hermanos (dos hombres y dos mujeres) integrantes de la misma unidad familiar y pertenecientes a la comunidad gitana. No han demandado atención sanitaria directa relacionada con esta anomalía.
Ambos padres de los sujetos del caso han fallecido. Ninguno manifestó fenotípicamente esta característica. Autoidentificados también como componentes de la población gitana, siendo un dato significativo la relación de parentesco –primos hermanos- entre ambos progenitores. De su unión nacieron los cuatro casos.
Desconocemos al padre, que murió a los 25 años cuando los hijos eran pequeños; la madre corroboró que su esposo no tenía esta particularidad. Según ella refiere, era delgado y de talla alta.
La madre tenía talla de 146 cm y peso de 83 kg (IMC: 38,96 % antes de enfermar). Murió en 2011 a los 45 años por cáncer de cávum. Tenía antecedentes de hipertensión arterial e hipotiroidismo subclínico.
La abuela materna de los casos estudiados, a la que llegamos a conocer, tampoco presentaba físicamente esta anomalía ni alteración visible que pudiera aludir a malformación genética a excepción de su talla baja: 145 cm.
Se obtuvo el consentimiento informado oral de los cuatro pacientes, tanto para las imágenes como para el uso del material. Su identidad se ha mantenido anonimizada, trabajando con la codificación de los casos. Sus datos se han protegido conforme a la Ley Orgánica de Protección de Datos (LOPD) de carácter personal 15/1999, al Real Decreto (RD) 1720/2007 y a la Ley 41/2002.
Los cuatro pacientes son adultos, presentan un desarrollo físico normal sin anomalías morfológicas aparentes salvo la particularidad referida. Todos han cursados estudios básicos primarios, alguno inacabado, sin evidencia de deterioro del desarrollo cognitivo, físico o psicomotor.
No existe asociación visible de la braquidactilia con otras afecciones, se han descartado talla baja, asimetrías, alteraciones genitales, fenotipos normalizados, o sea, que presentan rasgos normales sin otras alteraciones genéticas. A lo largo del tiempo se han ido evidenciando algunas anomalías, suponemos que están relacionadas con la manifestación sindrómica endocrina ya que ninguno de los casos ha sido estudiado.
Tres de los cuatro hermanos presentan obesidad, que en los casos 1 y 3 es mórbida y en el caso 2 es obesidad por encima del 30 %; los cuatro presentan hipotiroidismo, síndrome adaptativo y/o síndrome ansioso depresivo. Ninguno de ellos ha sido diagnosticado ni estudiado por la anomalía fenotípica o por aspectos relacionados con la misma. Todos tienen descendencia, sin evidencia física en ninguno de los descendientes de esta característica.
Asimismo, todos presentan una situación sociofamiliar similar, con escasez de recursos, marginalidad, exclusión social y dependencia económica basada, casi exclusivamente, en la ayuda institucional. Viven en un barrio marginal y en condiciones de infravivienda. Todos han manifestado que su anomalía no les ha impedido llevar a cabo las actividades habituales de la vida diaria, aunque han informado cierta vergüenza por su particularidad y tienden a esconder las manos. Su actividad profesional no está definida.
Narran que siempre les ha preocupado que sus descendientes presentaran dicha peculiaridad, y lo primero que querían descartar cuando han tenido una gestación era si los fetos tenían esa característica.
La malformación de los dedos de los pies no los ha afectado en el mismo sentido que la de las manos.
Los cuatro individuos presentan la manifestación de braquidactilia expresada fenotípicamente, pero con alguna variabilidad. En el caso 1 (figura 1), los datos radiológicos muestran una diferencia con respecto a los casos 2, 3 y 4: el acortamiento del tercer metacarpiano no encaja en el diagnóstico de braquidactilia tipo A4, por lo que se lo considera como una posible mezcla de A4 con E, o quizás como una nueva variedad no clasificada, ni registrada entre las que se encuentran en fase de clasificación (A5 y A6, posible A7). Los datos radiológicos evidencian que los casos 2, 3 y 4 (figura 2) corresponden a la braquidactilia tipo A4.
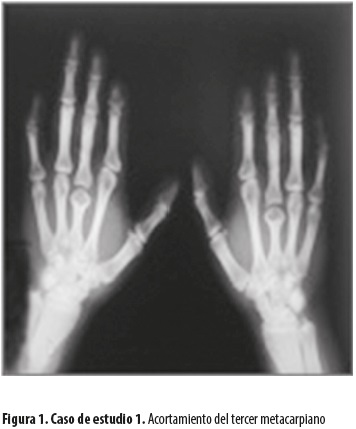
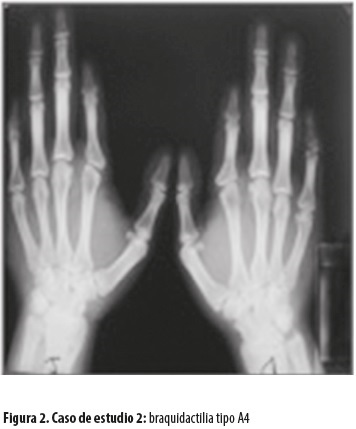
Los cuatro pacientes presentan anomalías similares en los pies, consistentes en displasia de las falanges distales de los dedos de ambos pies, generalmente el tercero y cuarto dedos; en tres de los casos son bilaterales y en uno, del pie izquierdo.
REPORTE DE LOS CASOS
Caso 1
Mujer de 32 años que presenta el rasgo descrito de braquidactilia que difiere del resto de los hermanos (figura 3). Casada a los catorce años y el marido a los diecisiete; tuvo su primera gestación a los 15 años, que acabó en aborto espontáneo en el primer trimestre del embarazo, y lo mismo ocurrió en la segunda gestación. Posteriormente, cuando contaba 16 años una gestación a término dio lugar al nacimiento de un varón. A continuación se produjeron dos abortos espontáneos más durante el primer trimestre de gestación. Del siguiente embarazo viable nació otro varón en 2003. Tras este parto se originó un nuevo embarazo cuando la paciente refería estar en tratamiento con anticonceptivos orales (ACO); al igual que en casos anteriores, este finalizó en aborto espontáneo también sin alcanzar los tres meses. Se ha desconocido siempre el sexo de los fetos abortados. El embarazo siguiente evolucionó hasta el sexto mes de gestación y concluyó con un aborto terapéutico por muerte fetal intrauterina (una niña) en 2006 cuando la mujer tenía veintitrés años.

Desde la primera gestación presentaba hipotiroidismo no tratado y aumento progresivo de peso que a lo largo de los años llegó a la obesidad. Dados los antecedentes obstétricos actualmente es portadora de DIU hormonal, según refiere por indicación médica, y manifiesta el deseo de aumentar su descendencia. Historia obstétrica patológica con dificultad para mantener la gestación a término. No había sido estudiada por estos motivos. No refiere relación de parentesco con su marido, pero sí cierta relación de parentesco lejano de sus padres con los padres de su marido (sus padres eran primos segundos). A su primer hijo varón se le diagnosticó trastorno de la coagulación Factor VIII cuando tenía cinco años y previa malformación bilateral del conducto auditivo intervenida y con hipoacusia. Peso 110 kg, talla 160 cm, IMC 42,96
Caso 2
Varón de 31 años que presenta el rasgo descrito de braquidactilia tipo A4, de similares características radiológicas a las de los otros dos hermanos menores. Casado en dos ocasiones, sin descendencia en la primera relación; de la segunda nació un hijo varón. Refiere relación lejana de parentesco con su mujer por parte de su padre. Ella también pertenece a la comunidad gitana. Tendencia a la obesidad desde temprana edad, debut relativamente reciente de hipotiroidismo, dislipidemia y trastorno adaptativo que obstaculiza las relaciones sociolaborales. Peso 105 kg, talla 178 cm, IMC 33,1.
Caso 3
Mujer de 28 años con la misma anomalía de sus hermanos, braquidactilia tipo A4 (figura 4). Casada a los dieciséis años, el marido a los veintitrés. Primera gestación a los 17 años, sin anomalías y a término con nacimiento de una niña. Esta ha sido su única gestación. Hasta el embarazo era delgada, aún no había debutado la clínica tiroidea, posteriormente ha presentado dificultades para concebir. Desde el parto mantiene un aumento progresivo de peso. Se detectó hipotiroidismo tras ablación tiroidea por bocio hipertiroideo; obesidad y dislipidemia, además de amenorrea. No refiere relación de parentesco con el padre de su hija, el cual informa no pertenecer a la población gitana. La hija nacida de esta relación presenta ostium secundum y está pendiente de cirugía cardíaca. Peso 125 kg, talla 177 cm, IMC 39,89.

Caso 4
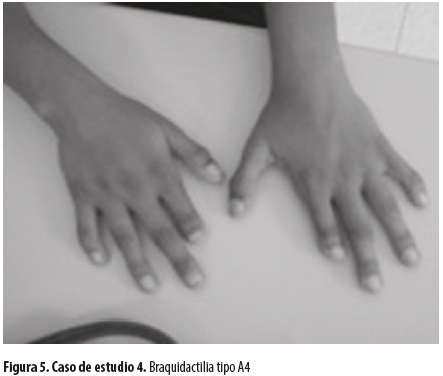
Varón de 26 años que, al igual que sus hermanos, presenta braquidactilia tipo A4 (figura 5) que no ha sido estudiada. Tiene problema de salud relacionado con estado ansioso depresivo. Casado con mujer perteneciente también a la comunidad gitana, sin relación de parentesco con ella. Tienen tres hijos: dos varones y una niña afectada de parálisis cerebral infantil. Peso 75 kg, talla 179 cm, IMC 23,40.
DISCUSIÓN
El primer rasgo que llamó nuestra atención y motivó este trabajo, aunque no el único, fue la circunstancia de que los cuatro hermanos manifestasen la misma anomalía. Es sorprendente que no hayan demandado asistencia sanitaria por esta peculiaridad, así como que desde el punto de vista sanitario, cuando han sido atendidos no se relacionase la alteración de la salud con esta singularidad. La braquidactilia, una enfermedad rara, que afecta a cuatro miembros de una misma familia y que no genera un diagnóstico, ni un interés por indagar en ella o en las posibles repercusiones sobre la salud de los afectados o de sus descendientes, requiere análisis y reflexión sobre el caso. En primer lugar, obliga a plantear la situación de que determinados individuos están infradiagnosticados y por tanto no ''tratados'' y habría que preguntarse el porqué de esta situación y con qué estaría relacionada. Cabe sugerir si la circunstancia de pertenecer a un colectivo culturalmente diferenciado como la población gitana y en situación de vulnerabilidad social, justificaría el desinterés del sistema sanitario y sus profesionales por esta familia (14).
Por otro lado, es posible que familias en situaciones de marginalidad, con bajo poder adquisitivo, graves dificultades en el acceso al mercado laboral, sin cualificación profesional, con condiciones precarias de vida y una anomalía morfológica, no se consideren interesantes y por tanto no generen una llamada de atención (15,16).
Nos preguntamos por qué no se ha planteado la necesidad de indagar en una posible malformación genética manifestada, aunque no demande atención sociosanitaria específicamente. La connotación de que afecte a una población culturalmente diferenciada, implícitamente parece contemplarse más como diferencia cultural entre grupos que como una situación de desigualdad o diferencias en salud de unos colectivos respecto a otros (17).
No podemos valorar sin un estudio previo en qué medida podría manifestarse en la siguiente generación una alteración genética, cuando la población presenta patrones de endogamia, consanguinidad y una situación socioambiental desfavorable. A la luz de los datos cabe pensar que quizás un estudio y consejo genéticos podrían haber amortiguado en cierta medida la situación sanitaria de estas cuatro familias y prevenir los procesos de morbimortalidad en los afectados y/o en sus descendencias.
Para empezar, pensamos que el estudio de la herencia genética debe constituir una condición básica en poblaciones consanguíneas o con altos índices de consanguinidad, ya que puede determinar desde la viabilidad hasta la morbimortalidad de la descendencia (18).
Por otro lado, que abordáramos el caso de forma conjunta no implica que los hallazgos en cada uno de los individuos sean idénticos, ya que las diferencias observadas en la exploración radiológica indican algunas variaciones; los casos 2, 3 y 4 son los más semejantes.
Centrándonos en primer lugar en la braquidactilia del caso 1, caracterizada por el acortamiento del tercer metacarpiano, en principio discreparía con el diagnóstico de braquidactilia tipo A4, pasando a ser una especie de mezcla de los tipos A4 y E, o, quizás, una nueva variedad no catalogada, ni registrada todavía entre las que están en fase de clasificación (A5 y A6, posible A7).
Ya hemos descrito que fenotípicamente no presentan otro rasgo diferenciador que el señalado, pero sí se han ido evidenciando otros problemas de salud o alteraciones en el desarrollo de los sujetos del estudio y parece que no se han relacionado con esta anomalía al no existir el estudio correspondiente.
Se evidencia cierto desinterés clínico por el caso 1, una mujer con dificultades para el mantenimiento de la gestación y con un historial obstétrico fallido no investigado. Cada gestación supone un riesgo para la salud de la mujer por la falta de diagnóstico.
Otros estudios ya han relatado gestaciones fallidas previas, en progenitoras aparentemente sin expresión fenotípica de esta anomalía genética (19), y que posteriormente han concebido con resultado de descendencia afectada (20).
Pensamos, sin embargo, que la expresión de ciertas anomalías genéticas puede hacer que pasen inadvertidas otras posibles alteraciones latentes; por tanto, cabe cuestionarse en qué condiciones se recurre al asesoramiento genético para abordar la transmisión y el desarrollo de trastornos hereditarios (21).
Es posible que determinadas situaciones de marginalidad dificulten el acceso a los recursos sanitarios, así como que aspectos culturales diferenciados y desconocidos por los profesionales condicionen que no sea adecuada la atención sociosanitaria a los individuos (22).
Sería preciso indagar si determinadas anomalías genéticas tienen diferentes frecuencias de aparición en los distintos grupos culturales, e intentar relacionar con los factores que podrían constituir rasgos culturales o si, por el contrario, se relacionan con otras variables y/o con diferencias en salud. Cabe pensar que la consanguinidad puede constituir un factor de riesgo añadido para el padecimiento o manifestación de determinadas anomalías, en algunos casos con graves consecuencias para la salud. Es posible, como en el caso que nos ocupa, que la braquidactilia con patrón de herencia recesiva, se transforme a dominante, cuando concurren situaciones de endogamia y consanguinidad, aumentando por tanto la probabilidad de su presentación. Es decir, que exista una mayor probabilidad de presentar más anomalías o malformaciones y por tanto haya mayor probabilidad de morbimortalidad.
Es preciso mejorar los mecanismos de información y asesoramiento genético a colectivos en que la consanguinidad constituye un rasgo cultural; es preciso también incidir en los posibles riesgos derivados de esta circunstancia, tanto para ellos mismos como para la descendencia, en cuanto a determinadas afecciones y de las posibles alternativas existentes (23). En definitiva, atender a las necesidades específicas de este colectivo, lo que incluye aspectos culturales y adaptar los servicios sanitarios a la diversidad social existente (24).
Que se centre la atención de estos casos expuestos en aspectos concretos como la obesidad, los síndromes adaptativos, etc., quizás evidencia un abordaje inadecuado de la situación, pues implícitamente descargan o justifican la situación de los sujetos en aspectos personales vinculados a hábitos, costumbres, capacidad de afrontamiento, conductas diferenciadas que ''justificarían'' la patología, pues determinados estilos de vida serían electivos, y por tanto justificarían la enfermedad, la marginalidad y la exclusión social (25-27).
En el caso que nos ocupa, que los pacientes no mantengan una conducta de cumplimiento terapéutico constituye solo la parte visible del iceberg. Sabemos que la adhesión al régimen terapéutico en cierta medida está condicionada por creencias, costumbres, necesidades, la capacidad para afrontar el gasto sanitario que supone, por la prioridad en la satisfacción de necesidades básicas más importantes que las del tratamiento farmacológico, como vivienda, comida, bienestar de los hijos etc., (28,29), por la confianza o no en el sistema sanitario, relacionada con expectativas respecto a este, alternancia con otras prácticas y alternativas en el mantenimiento y recuperación de la salud (30,31)
En definitiva, cabe pensar que en la agregación de variables que determinan un empeoramiento del problema de salud concurren las situaciones relacionadas con la marginalidad, la exclusión y la pobreza, que condicionan la morbilidad y se proyectan sobre el propio individuo responsabilizándolo de su situación, mientras se justifican las instituciones y profesionales ante la incapacidad para atender a la diversidad desde el punto de vista sanitario y el acceso desigual a los recursos de unos individuos frente a otros.
CONCLUSIÓN
La consanguinidad se relaciona con un peor estado de salud al aumentar el riesgo de morbimortalidad para determinadas afecciones como es la braquidactilia en este caso. Sin embargo, existen otras variables como marginalidad, pobreza, déficit de recursos y acceso desigual a ellos, que inciden de igual manera y/o condicionan la expresividad de dichas alteraciones y no se están considerando ni interpretando en la misma clave de riesgo.
En población con concurrencia de factores adversos, la salud de los individuos tiende a seguir el mismo patrón, las bolsas de pobreza generan también bolsas de enfermedad. Es imprescindible acercar los recursos sanitarios a la población. El desarrollo y avance científicos deben redundar en todos los colectivos sociales, atendiendo a las necesidades específicas en salud de los mismos. Sin embargo, no será suficiente si no se acompañan de medidas estructurales que afecten al ámbito social, educativo, económico, político y cultural de la sociedad y especialmente de los colectivos más desfavorecidos.
REFERENCIAS BIBLIOGRÁFICAS
1. Morar B, Gresham D, Angelicheva D, Tournev I, Gooding R, Guergueltcheva V, et al. Mutation history of the roma/gypsies. Am J Hum Genet. 2004 Oct;75(4):596-609. [ Links ]
2. Robson AG, Webster AR, Michaelides M, Downes SM, Cowing JA, Hunt DM, et al. ''Cone dystrophy with supernormal rod electroretinogram'': a comprehensive genotype/phenotype study including fundus autofluorescence and extensive electrophysiology. Retina. 2010 Jan;30(1):51-62. DOI 10.1097/IAE.0b013e3181bfe24e. [ Links ]
3. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010 Apr;7(4):248-9. DOI 10.1038/nmeth0410-248. [ Links ]
4. Chamova T, Florez L, Guergueltcheva V, Raycheva M, Kaneva R, Lochmüller H, et al. ANO10 c.1150_1151del is a founder mutation causing autosomal recessive cerebellar ataxia in Roma/Gypsies. J Neurol. 2012 May;259(5):906-11. DOI 10.1007/s00415-011-6276-6. [ Links ]
5. Holtzman NA, Marteau TM. Will genetics revolutionize medicine? N Engl J Med. 2000 Jul;343(2):141-4. [ Links ]
6. Bottorff JL, Blaine S, Carroll JC, Esplen MJ, Evans J, Nicolson Klimek ML, et al. The educational needs and professional roles of Canadian physicians and nurses regarding genetic testing and adult onset hereditary disease. Community Genet. 2005;8(2):80-7. [ Links ]
7. Gamella JF, Carrasco-Muñoz EM, Núñez Negrillo AM. Oculocutaneous albinism and consanguineous marriage among Spanish Gitanos or Calé--a study of 83 cases. Coll Antropol. 2013 Sep;37(3):723-34. [ Links ]
8. Dathe K, Kjaer KW, Brehm A, Meinecke P, Nürnberg P, Neto JC, et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am J Hum Genet. 2009 Apr;84(4):483-92. DOI 10.1016/j.ajhg.2009.03.001. [ Links ]
9. Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW, et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet. 2007 Aug;81(2):388-96. [ Links ]
10. Demirhan O, Türkmen S, Schwabe GC, Soyupak S, Akgül E, Tastemir D, et al. A homozygous BMPR1B mutation causes a new subtype of acromesomelic chondrodysplasia with genital anomalies. J Med Genet. 2005 Apr;42(4):314-7. [ Links ]
11. Plöger F, Seemann P, Schmidt-von Kegler M, Lehmann K, Seidel J, Kjaer KW, et al. Brachydactyly type A2 associated with a defect in proGDF5 processing. Hum Mol Genet. 2008 May;17(9):1222-33. DOI 10.1093/hmg/ddn012. [ Links ]
12. Kjaer KW, Eiberg H, Hansen L, van der Hagen CB, Rosendahl K, Tommerup N, et al. A mutation in the receptor binding site of GDF5 causes Mohr-Wriedt brachydactyly type A2. J Med Genet. 2006 Mar;43(3):225-31. [ Links ]
13. Temtamy SA, Aglan MS. Brachydactyly. Orphanet J Rare Dis. 2008 Jun;3:15. DOI 10.1186/1750-1172-3-15. [ Links ]
14. Gracia JN, Ruffin J. Partnership, research, and leadership to advance health equity and eliminate health disparities. Am J Public Health. 2014 Sep;104 Suppl 4:S520-1. DOI 10.2105/AJPH.2014.302201. [ Links ]
15. Clark C. Defining ethnicity in a cultural and sociolegal context: the case of Scottish Gypsy-Travellers. Scottish Affairs. 2006;54:39-67. [ Links ]
16. Karlsen S, Nazroo JY. Agency and structure: the impact of ethnic identity and racismo on the health of ethnic minority people. Sociol Health Ill. 2002;24(1):1-20. [ Links ]
17. La Parra D, Gil-González D, Jiménez A. Los procesos de exclusión social y la salud del pueblo gitano en España. Gac Sanit. 2013 Sep-Oct;27(5):385-6. DOI 10.1016/j.gaceta.2013.05.001. [ Links ]
18. Wade C, Brinas I, Welfare M, Wicking C, Farlie PG. Twist2 contributes to termination of limb bud outgrowth and patterning through direct regulation of Grem1. Dev Biol. 2012 Oct;370(1):145-53. DOI 10.1016/j.ydbio.2012.07.025. [ Links ]
19. Cervantes-Barragán DE, Villarroel CE, Medrano-Hernández A, Durán-McKinster C, Bosch-Canto V, Del- Castillo V, et al. Setleis syndrome in Mexican-Nahua sibs due to a homozygous TWIST2 frameshift mutation and partial expression in heterozygotes: review of the focal facial dermal dysplasias and subtype reclassification. J Med Genet. 2011 Oct;48(10):716-20. DOI 10.1136/jmedgenet-2011-100251. [ Links ]
20. Chaabouni M, Le Merrer M, Raoul O, Prieur M, de Blois MC, Philippe A, et al. Molecular cytogenetic analysis of five 2q37 deletions: refining the brachydactyly candidate region. Eur J Med Genet. 2006 May-Jun;49(3):255-63. [ Links ]
21. Foster MW, Sharp RR. Race, ethnicity, and genomics: social classifications as proxies of biological heterogeneity. Genome Res. 2002 Jun;12(6):844-50. [ Links ]
22. Flodgren G, Parmelli E, Doumit G, Gattellari M, O'Brien MA, Grimshaw J, et al. Local opinion leaders: effects on professional practice and health care outcomes. Cochrane Database Syst Rev. 2011 Aug;(8):CD000125. DOI 10.1002/14651858.CD000125.pub4. [ Links ]
23. Bergeron DA, Bourgault P, Gallagher F. [Nursing activities in family medicine groups for patients with chronic pain]. Pain Res Manag. 2015 Mar-Apr;20(2):101-6. [ Links ] French.
24. Conn VS, Enriquez M, Ruppar TM, Chan KC. Cultural relevance in medication adherence interventions with underrepresented adults: systematic review and meta-analysis of outcomes. Prev Med. 2014 Dec;69:239-47. [ Links ] DOI 10.1016/j.ypmed.2014.10.021.
25. Conway SM. Health education: leading the way to a healthy future. NASN Sch Nurse. 2015 Jan;30(1):10-2. [ Links ]
26. Speroni KG. School nurse facilitated programs for families living fit. NASN Sch Nurse. 2014 May;29(3):140-4. [ Links ]
27. Bedford S, Jones E. Should lifestyle choices affect access to transplant? Nurs Times. 2014 Jul;110(30):16-8. [ Links ]
28. Rathwallner B. [The family health nurse concept. Nursing care at the center of society]. Pflege Z. 2014 Nov;67(11):660-2. [ Links ] German.
29. Salomé GM, de Almeida SA, Ferreira LM. Association of sociodemographic factors with hope for cure, religiosity, and spirituality in patients with venous ulcers. Adv Skin Wound Care. 2015 Feb;28(2):76-82. DOI 10.1097/01.ASW.0000459844.07689.02. [ Links ]
30. Schorn MM, Doorenbos AZ, Gordon D, Read-Williams P. Survey of Primary-Care Providers on Perceived Benefits of and Barriers to PainTracker. J Nurse Pract. 2014 Dec;10(10):781-786. [ Links ]
31. Jongudomkarn D, Macduff C. Development of a family nursing model for prevention of cancer and other noncommunicable diseases through an appreciative inquiry. Asian Pac J Cancer Prev. 2014;15(23):10367-74. [ Links ]

