











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome oro-facio-digital (OFD) es un trastorno complejo del desarrollo, descrito en 1941 por el genetista noruego Otto J. Mohr quien reportó por primera vez un caso con las manifestaciones clínicas de la variante tipo II1; posteriormente, en 1954, en Francia, los dentistas Papillón-Lèage y Psaume describieron la variante tipo I1)(2. Su incidencia mundial es baja por lo que se incluye entre las enfermedades raras3. Los síndromes oro-facio-digitales son un grupo heterogéneo de trastornos en los que se han descrito al menos catorce formas diferentes de manifestarse4. El síndrome OFD II o síndrome de Mohr se transmite como un trastorno autosómico recesivo caracterizado por malformaciones de la cavidad oral, la cara y los dedos. Entre las características faciales y orales cabe destacar la aparición de nódulos en la lengua, fisura labio/palatina, agenesia dental y nariz ancha. Las características de los dedos incluyen clinodactilia, polidactilia, sindactilia, braquidactilia y duplicación del pulgar. Se han descrito además sordera conductiva, coloboma de la coroides y defectos congénitos cardíacos y renales. El diagnóstico se basa en las características clínicas5.
En Colombia se han informado pocos casos de este síndrome, por lo que se ignoran sus implicaciones epidemiológicas. Se presenta y discute el caso de un niño con diagnóstico clínico de OFD.
PRESENTACIÓN DEL CASO
Niño de 10 años de edad, procedente de Bogotá, Colombia, escolar de cuarto de primaria, con diagnóstico presuntivo de síndrome oro-facio-digital, que consultó al Grupo de Genética Humana en la Fundación Operación Sonrisa Colombia después de la palatorrafia. Fue evaluado por el servicio de Otorrinolaringología donde le diagnosticaron hipoacusia grave bilateral que requirió implante coclear con mejoría de la audición; fue producto del tercer embarazo postérmino y nació por cesárea debido a preeclampsia y diabetes gestacional, con peso y talla al nacer adecuados para la edad gestacional. Desarrollo psicomotor adecuado para la edad. Ha requerido queilorrafia, palatorrafia y herniorrafia inguinal izquierda, miringostomía e implante coclear bilateral. Niega consanguinidad paterna. Antecedentes familiares: una tía materna con fisura labio-palatina y un primo materno con hiperlaxitud articular. Otros antecedentes fueron negativos.
Los hallazgos al examen físico fueron los siguientes: talla para la edad entre el percentil (p) 0 a -1 (adecuada); peso para la edad entre p: 0 a -1 (adecuado); perímetro cefálico p: +2 (macrocefalia)6; distancia intercantal interna: 97 %; distancia intercantal externa: 97 % (hipertelorismo y telecanto secundario)7; raíz nasal alta, fisura labio-palatina al lado derecho corregida, paladar sin fístulas, micrognatia leve (Figura 1), pectus excavatum asimétrico derecho, hiperlordosis leve (Figura 2), ruidos cardíacos rítmicos con soplo sistólico aórtico, clinodactilia bilateral del quinto dedo, hiperlaxitud en las manos, cubitus valgus, talo valgo bilateral, sindactilia bilateral del segundo y tercer dedos (Figura 3), leve asimetría en la longitud de los miembros inferiores (Figura 4), el resto del examen físico fue normal.
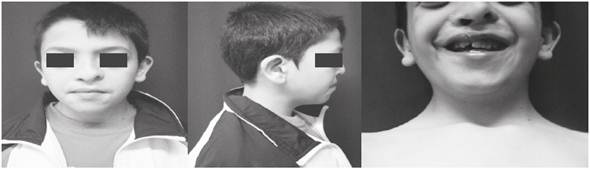
Figura 1 Fenotipo oro-facial. A) asimetría facial, hipertelorismo, raíz nasal alta, cicatriz de corrección de la fisura labial. B) Micrognatia. C) Cicatriz de corrección de la fisura y aglomeración dental
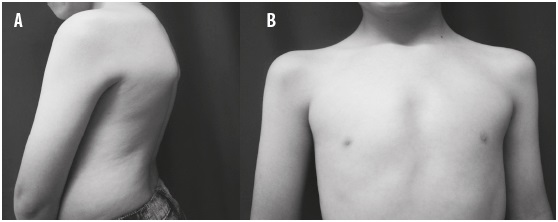
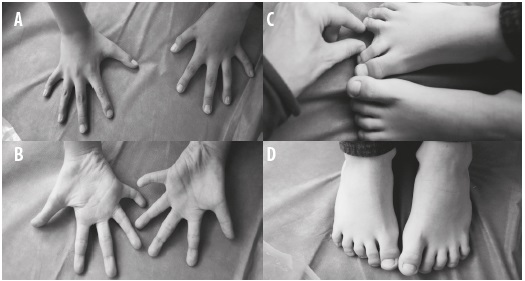
Figura 3 Fenotipo digital de las manos y los pies. A y B) Manos: clinodactilia bilateral en los meñiques, sindactilia bilateral del segundo y tercer dedos e hiperlaxitud. C y D) Pies: hiperlaxitud
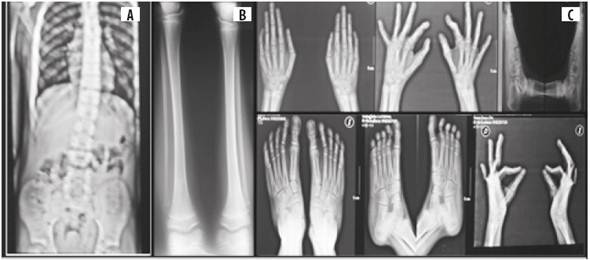
Figura 4 Fenotipo radiológico. A) Hiperlordosis y asimetría del tórax. B) Leve asimetría de los miembros inferiores y talo valgo bilateral. C) clinodactilia bilateral de los meñiques y sindactilia bilateral del segundo y tercer dedos
Radiología: en la radiografía de los huesos largos se evidenciaron sindactilia cutánea en la falange proximal de los meñiques, sindactilia bilateral del segundo y tercer dedos, asimetría de los huesos largos (0,5 cm); en la radiografía de tórax se halló hiperlordosis leve (Figura 4). Por los hallazgos clínicos y radiológicos se diagnosticaron síndrome OFD, hipoacusia mixta con implante coclear y displasia esquelética.
DISCUSIÓN
El síndrome oro-facio-digital (OFD) es una alteración congénita infrecuente caracterizada por alteraciones faciales, de la cavidad bucal y dactilares3; también se han informado, entre otras, alteraciones renales, óseas y del canal auditivo3)(4)(7)(8)(9)(10)(11)(12)(13. Hasta el momento se han descrito 14 variantes3, cada una caracterizada por diferentes alteraciones fenotípicas y genotípicas. La prevalencia difiere según la variante: la más común (1/250 000 a 1/500 000 nacidos vivos) es el tipo I, mientras que la de las otras 14 variantes se calcula en menos de 1/1 000 000 de nacidos vivos8)(9)(10. No se dispone en Colombia de datos epidemiológicos específicos sobre esta enfermedad.
La mayoría de los casos son familiares con patrón de herencia autosómico recesivo; sin embargo, en el OFD I la herencia está ligada a X dominante14)(15. La causa del OFD es una mutación genética, pero hasta ahora solo se ha establecida la de la variante tipo I, que es una mutación única en el gen CXORF5, localizado en el brazo corto del cromosoma X (Xp22.3- 22.2), que codifica para la producción de la proteína OFD I, esencial para la supervivencia fetal y el desarrollo temprano de todos los órganos15. Todas las variantes comparten alteraciones físicas lo que dificulta el diagnóstico diferencial.
El síndrome oro-facio-digital tipo II o síndrome de Mohr tiene herencia autosómica recesiva; se caracteriza por un fenotipo de estatura corta, hipoacusia conductiva, hipertelorismo y/o telecanto, el puente nasal puede ser bajo, la punta nasal puede ser ancha y/o bífida. En la boca hay múltiples alteraciones, las más comunes son: fisura labio/palatina, lengua lobulada o con nódulos y ausencia del incisivo central. Otras alteraciones posibles son: pectus excavatum, escoliosis, hipoplasia maxilar y del arco zigomático, irregularidades metafisiales, manos con clinodactilia, braquidactilia y/o polidactilia bilateral, en los pies puede haber polidactilia postaxial, duplicación del hallux, hueso cuneiforme extra y cuboides amplio en el primer metatarsiano. Con respecto al sistema nervioso central, se pueden presentar hidrocefalia y/o porencefalia, pero la mayoría de estos pacientes tienen nivel intelectual normal9. Con base en las características clínicas del paciente y la información disponible sobre la variante de OFD tipo II se hizo el diagnóstico de esta variante, al evidenciar la ausencia de otras manifestaciones clínicas tales como cerebrales, renales, esqueléticas, extradigitales, de la vía aérea, oculares, genitales o neuropsiquiátricas. Se excluyeron los tipos I y III a XIV.
Aunque la hipoacusia y la fisura labio-palatina también se presentan en la variante tipo VI, el paciente reportado no tenía nistagmus, agenesia renal, ni déficit cognitivo11. También hay características que comparte el paciente con la variante tipo IV: el pectus excavatum, el labio hendido y las alteraciones en manos y pies, pero en este tipo no hay hipoacusia ni alteraciones nasales12 que el paciente descrito sí presentaba. La variante tipo VIII, que comparte con el paciente descrito el hipertelorismo, las alteraciones nasales y la fisura labio-palatina, se puede excluir por la ausencia de antecedentes de neumonías a repetición y de alteraciones epigástricas13.
La morbimortalidad y el pronóstico de los pacientes con OFD dependen de las manifestaciones clínicas específicas y de su gravedad; en el paciente descrito lo más relevante fue la hipoacusia que requirió un implante coclear; para orientar los estudios diagnósticos es importante identificar la variante sindrómica; en el tipo II se deben hacer estudios de potenciales evocados auditivos, tomografía axial computarizada o resonancia magnética nuclear cerebral y radiografías de huesos largos, de manos y pies.
El reporte del caso de este paciente con OFD tipo II es importante por su infrecuencia y porque se desconoce el gen específico implicado. Se lo puede incluir en la casuística de informes basados en el diagnóstico clínico, para estudios de búsqueda del gen causal con las nuevas tecnologías genómicas y de esta forma garantizar un diagnóstico acertado y mejorar el pronóstico. Además, es importante que los médicos generales y los pediatras conozcan este síndrome porque en la actualidad el diagnóstico es solamente clínico.

