











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
Los errores innatos del metabolismo (EIM) son un área de estudio en continuo crecimiento. En los últimos 10 años el número de EIM informados ha pasado de 500 a aproximadamente 1000, lo que plantea a los médicos los desafíos de adquirir nuevos conocimientos y de tener en cuenta, entre las posibilidades diagnósticas, enfermedades poco conocidas e infrecuentes. Al ampliar la información científica, se producen avances en el diagnóstico y tratamiento de estas enfermedades y crece el interés por su detección precisa y oportuna. Esto puede plantear el riesgo de desviar el curso del diagnóstico de enfermedades con alta prevalencia, e incluso puede llevar a que el profesional genere expectativas diagnósticas y terapéuticas para el paciente, la familia y el personal de la salud.
Presentamos el caso de una paciente con historia de noxa perinatal y alteraciones neurológicas a quien le hicieron múltiples estudios para EIM, pero cuyo curso clínico no mostró la progresión típica de las enfermedades metabólicas; desde el comienzo se evidenciaron en la resonancia magnética cerebral lesiones glióticas en la sustancia blanca compatibles con un evento antiguo de posible origen hipóxico-isquémico.
PRESENTACIÓN DEL CASO
Lactante de sexo femenino traída a la consulta de neurología pediátrica, pero atendida desde los 9 meses en la de pediatría; estaba en estudio por presentar crisis de espasmos infantiles, correspondientes al síndrome de West. Es producto del primer embarazo, de padres no consanguíneos, se documentó restricción del crecimiento intrauterino (RCIU) mediante ecografía obstétrica a las 30 semanas. Parto por cesárea a las 36 semanas de gestación por preeclampsia detectada a las 34 semanas. Peso al nacer 2200 gramos, talla 41cm. Las pruebas de tamizaje materno para TORCH fueron negativas. Tuvo episodios de apnea que se resolvieron. No se tienen datos del APGAR. Recibió fototerapia durante 2 días por ictericia fisiológica. La madre y dos tíos maternos tienen síndrome de EhlersDanlos tipo IV (forma vascular), que fue monitorizado durante la gestación, pero no hubo cardiopatía materna.
El desarrollo psicomotor estuvo alterado desde muy temprano, con sostén cefálico parcial a los 3 meses, cambio de posición supina a prona a los 9 meses, se sostiene sentado sin apoyo a los 17 meses, pero no ha logrado la marcha. Lenguaje: monosílabos. Al cuarto mes de edad presentó espasmos infantiles y se inició tratamiento con piridoxina y vigabatrín; ha tenido múltiples hospitalizaciones, mal control de las crisis epilépticas y presencia de otras crisis como ausencias, caída cefálica y focales motoras, por lo que fue necesario hacer ajustes en el tratamiento con vigabatrín, clobazam, ácido valproico y levetiracetam, con los que se controlaron los espasmos. A los 10 meses de edad no se evidenciaron en el examen alteraciones fenotípicas, organomegalias ni alteraciones óseas; perímetro cefálico 41cm (-3 DS), pares craneanos normales, reflejos musculotendinosos exaltados generalizados; sostén cefálico en posición prona inestable, no sedente, hipotonía generalizada. La paciente no ha presentado alteraciones sugestivas de un EIM del primer año de edad, a saber: cuadros de descompensación metabólica, alteraciones cardíacas o musculares, intolerancia alimentaria, compromiso visceral o hematológico u otras.
Los aminoácidos y los ácidos láctico y pirúvico en sangre y LCR fueron normales (Tabla 1). Pese al difícil control de sus crisis, ha tenido aumento del perímetro cefálico, a los 26 meses ya era de 46 cm (-1 DS), y adquisición de hitos del desarrollo, logrando a los 29 meses respuestas protectoras simétricas en posición sedente, postura de cuadrúpedo, bipedestación con apoyo, bisílabos con intención comunicativa. Las funciones auditiva y visual fueron normales. En las primeras imágenes de resonancia magnética cerebral (RMC) se observó leucoencefalopatía peri- y paraventricular y en los centros semiovales y dilatación leve del sistema ventricular, hallazgos interpretados como una posible leucodistrofia, por lo que se ampliaron los estudios en busca de enfermedad lisosomal. Se midieron los niveles de hexosaminidasas A y B, gangliósidos GM2 y beta-galactosidasa, para gangliósidosis GM1; y los de arilsulfatasas A (ARSA) y B, para leucodistrofia metacromática (LDM) y mucopolisacaridosis tipo VI (Tabla 2). Los niveles leucocitarios de ARSA estuvieron ligeramente disminuidos, pero dentro del rango de normalidad para la población general; se hicieron dos mediciones con resultados de 9,41 nmol/h/ mg y 8,11 nmol/h/mg (Tabla 2). Estas cifras equivalen a un porcentaje de actividad residual entre 30 % y 50 % del control normal. En general, se considera que las enfermedades de depósito lisosomal se expresan cuando la actividad residual de la enzima no supera el 10 % frente a los controles normales.
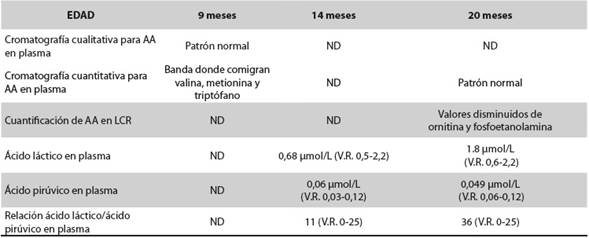
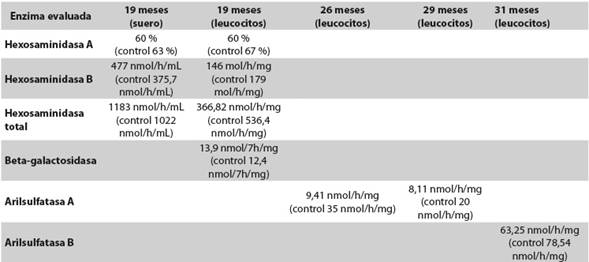
Las imágenes cerebrales, analizadas por un segundo neurorradiólogo, fueron informadas como sugestivas de noxa perinatal de posible origen hipóxico-isquémico; en los controles siguientes no se evidenció progresión de las lesiones, ni aparición de un patrón sugestivo de alguna de las leucodistrofias de origen metabólico (Figuras 1 y 2). La espectroscopia por RMC fue normal (Figura 3).
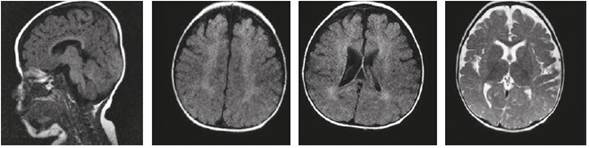
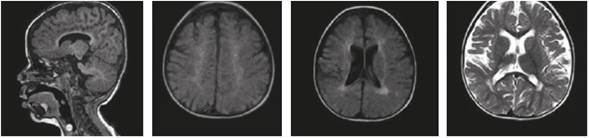
Figura 2 Resonancia magnética cerebral simple, a los 16 meses de edad. No se observan cambios compatibles con progresión
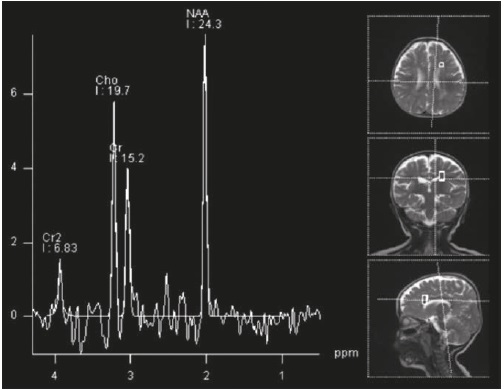
Se hicieron estudios adicionales para EIM del metabolismo intermedio y de molécula grande, cuyos resultados fueron negativos. Las Tablas 3 y 4 presentan los resultados de los demás exámenes. La paciente continúa en manejo interdisciplinario, con avances lentos en el neurodesarrollo, pero mal control de las crisis. Se inició dieta cetogénica.


DISCUSIÓN
El enfoque diagnóstico inicial de un EIM no difiere mucho del de cualquier enfermedad no metabólica; se debe iniciar en forma simultánea con la búsqueda de entidades de mayor incidencia, especialmente en el período neonatal. La historia familiar y los antecedentes perinatales son importantes para la sospecha clínica. En este caso, no hay historia de consanguinidad, abortos o muertes perinatales inexplicadas. La madre tiene enfermedad de Ehlers-Danlos vascular, que se ha descrito como causa de una mayor probabilidad de presentar lesiones en los tejidos blandos vaginales y perineales durante el parto debido a las alteraciones del colágeno propias de esta entidad1. Aún es controversial si durante la gestación esta enfermedad puede afectar directamente al feto, pero hay informes sobre un riesgo aumentado de prematuridad, aborto espontáneo, ruptura uterina y muerte materna2.
En el caso presente son llamativas el RCIU y la prematuridad, que pueden ser indicios de noxa previa para la enfermedad de la paciente; su origen es difícil de establecer en ausencia de infección o alteraciones genéticas, pero difiere del esquema clásico de la mayoría de los EIM, que usualmente cursan con parto a término y peso y talla adecuados al nacer3. A estos hechos se suma la preeclampsia materna con las complicaciones fetales asociadas, alteraciones en la microvasculatura e insuficiencia placentaria crónica evidenciada en el RCIU, activando mecanismos compensatorios en el flujo sanguíneo cerebral con prioridad para el tallo y los centros vitales sobre otras estructuras encefálicas como la corteza temporal y occipital, el hipocampo y el cerebelo4. Son materia de discusión las alteraciones del neurodesarrollo en los pacientes que presentaron RCIU, que han tenido resultados diferentes en el seguimiento a largo plazo4.
El cuadro clínico y el examen físico en los diferentes grupos etarios son importantes en el enfoque diagnóstico de los EIM3)(5; son hallazgos sugestivos las facies dismórficas, los olores inusuales, las alteraciones del sistema nervioso central como ataxia, extrapiramidalismo, neuropatía periférica, anormalidades visuales, auditivas, cardíacas y musculares, y el compromiso óseo y/o visceral; la presencia de cuadros intermitentes de toxicidad y acidosis metabólica asociados a falla en el medro, hipoglicemias e intolerancia alimentaria5)(6)(7. En el caso descrito, dada la ausencia de estos síntomas, conviene buscar otras causas de alteraciones neurológicas. La epilepsia es un elemento importante con curso de encefalopatía epiléptica, determinante en las alteraciones del neurodesarrollo. El patrón electroencefalográfico de hipsarritmia abre la posibilidad diagnóstica de un EIM, ya que puede estar asociado a entidades como hiperglicinemia no cetósica3; el síndrome de West, como ocurrió en este caso, puede ser parte de trastornos como deficiencia de piruvato-deshidrogenasa, piruvato-carboxilasa, defecto de glicosilación tipo III y aminoacidurias orgánicas. La paciente presentó perfil bioquímico normal y se descartaron trastornos del metabolismo intermediario; su cuadro clínico no es compatible con las enfermedades anotadas, la etiología más frecuente del síndrome de West es la hipoxia perinatal. La evolución clínica y electroencefalográfica de sus crisis epilépticas sugiere la transformación hacia el síndrome de Lennox-Gastau.
Las imágenes en la RMC cerebral dan información importante; las lesiones que alteran la sustancia blanca y que empeoran en controles sucesivos, unidas a regresión del neurodesarrollo, ayudan al diagnóstico clínico de leucodistrofia y hacen necesario hacer el diagnóstico diferencial con aquellas entidades asociadas a un defecto metabólico específico, y con las debidas a otras causas como las hipomielinizantes o las que presentan síntomas sistémicos característicos8)(9)(10. La paciente no presentó cuadro de regresión del neurodesarrollo, sino retraso psicomotor desde muy temprana edad que, pese al mal control de las crisis epilépticas, ha ido evolucionando hacia la adquisición lenta de hitos del desarrollo y no a la pérdida de los mismos como sucede en las enfermedades degenerativas de origen metabólico con afectación de la sustancia blanca. Existen leucodistrofias de presentación connatal, que son de curso progresivo, con manifestaciones visuales, auditivas y multisistémicas, pero esta no es una opción diagnóstica en la paciente. La ausencia de un patrón radiológico compatible con alguno de los EIM11)(12, así como el carácter estático de las lesiones que sugieren gliosis como resultado de un evento antiguo no progresivo, sumados a la espectroscopia normal por RMC, disminuyen la posibilidad de EIM en esta paciente.
En lo que respecta a los niveles de ARSA, como en la mayoría de las enfermedades de depósito lisosomal, se acepta que una deficiencia significativa, menor que el 10 % del control normal, asociada a un cuadro clínico compatible y a excreción de sulfátidos en la orina es indicativa de LDM5)(10; una deficiencia entre 15 % y 50 %, como ocurre con el caso en referencia, puede estar presente en familiares de pacientes afectados por LDM o incluso en individuos sanos13, que no presentan depósitos de material metacromático ni excretan sulfátidos en la orina. A esta condición se la conoce como pseudodeficiencia de arilsulfatasa A, y se la ha informado en cerca de 20 % de la población general. Estos individuos suelen ser asintomáticos, aunque ocasionalmente pueden padecer trastornos neuropsiquiátricos como autismo y alteraciones comportamentales y del aprendizaje14. En el caso presente, no fue posible hacer la determinación de sulfátidos en la orina para corroborar una pseudodeficiencia; sin embargo, la edad y la forma de presentación de su enfermedad no corresponden a una forma temprana ni juvenil de LDM.
Otra posible explicación para niveles ligeramente bajos de ARSA es la deficiencia del cofactor no enzimático de ARSA, el saposín B. La presentación clínica de la enfermedad por déficit de este cofactor es clínicamente muy similar a la LMD clásica, pero la actividad de ARSA es normal o está ligeramente disminuida; en estos casos el diagnóstico solo se puede establecer mediante estudio molecular del gen codificante para el saposín B. En el caso presentado las características clínicas no son compatibles con esta posibilidad15)(16.
En síntesis, se presenta la historia de una paciente con noxa perinatal, retraso global del desarrollo y alteraciones neurológicas, que no muestra curso involutivo ni afectación multisistémica, asociadas a lesiones en la sustancia blanca documentadas por RMC cerebral en la que no se observan cambios sugestivos de un proceso desmielinizante progresivo y que en cambio parecen compatibles con un evento antiguo de posible origen hipóxico-isquémico.
Debe aclararse que se logró analizar este caso gracias a los resúmenes de la historia clínica provenientes de distintos centros de atención y que fueron hechos cada vez por un especialista diferente, hecho frecuente en nuestro país debido a las condiciones propias del sistema de salud, pero que muchas veces contribuye al retraso del proceso diagnóstico y origina interpretaciones a menudo incompletas y confusas. En este caso, como en muchos otros, no hay continuidad durante el proceso diagnóstico, por lo que los profesionales de la salud fácilmente pueden omitir detalles de la historia clínica e incluso no tener conocimiento de las ayudas diagnósticas ya hechas, lo que en suma contribuye a la pérdida del flujo normal en el curso del enfoque diagnóstico.
CONCLUSIONES
Se resalta la necesidad de hacer el enfoque clínico partiendo de las enfermedades más frecuentes y conocidas, pero considerando al mismo tiempo la posibilidad diagnóstica de otras menos prevalentes y más complejas como los EIM, tratando en lo posible que el proceso diagnóstico sea interdisciplinario y continuo en beneficio del paciente. De igual manera, cabe resaltar la importancia de la semiología clínica en el momento del enfoque diagnóstico. El análisis detallado de la historia clínica y de los antecedentes familiares permite en la mayoría de los casos orientar el diagnóstico, que luego puede ser apoyado por las ayudas paraclínicas. Lo anterior es benéfico porque representa un ahorro en términos de tiempo y dinero al hacer uso racional de dichas ayudas y evitarle al paciente numerosas consultas, exámenes y trámites administrativos que impactan negativamente su calidad de vida.

