











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La enfermedad de Gaucher (EG) es un trastorno autosómico recesivo causado por la deficiencia de glucocerebrosidasa, los pacientes presentan gran variabilidad fenotípica y un amplio espectro de manifestaciones. Desde el punto de vista ocular, puede afectar estructuras tanto del segmento anterior como del posterior; las opacidades vítreas están entre las manifestaciones más llamativas. Presentamos el caso de una paciente con compromiso sistémico y ocular por esta enfermedad.
PRESENTACIÓN DEL CASO
Mujer de 15 años de edad, remitida desde su hospital local al Hospital Pablo Tobón Uribe (HPTU), Medellín, Colombia, por estado convulsivo. Desde los 8 años de edad ha presentado episodios convulsivos; ha estado en tratamiento con ácido valproico, pero no lo recibía desde una semana antes.
Luego de iniciar la escolaridad se evidenciaron problemas en el desarrollo pondoestatural y neurológico. Además, desde hace 2 años presenta epistaxis recurrentes y desde hace 4 meses, aumento del volumen abdominal.
Como antecedentes resaltan la consanguinidad entre los padres (primos hermanos), fractura de la cadera derecha a los 9 años de edad y un hermano en edad escolar con retraso en el neurodesarrollo.
Examen físico: facies dismórfica y marcada esplenomegalia. Se la hospitaliza en la unidad de cuidados intensivos con diagnóstico de trabajo de síndrome convulsivo secundario.
Exámenes paraclínicos: la ecografía abdominal muestra esplenomegalia masiva de 22 cm; en el hemograma se evidencia bicitopenia (trombocitopenia y anemia); en la resonancia magnética cerebral no hay alteraciones. El electroencefalograma mostró hallazgos sugestivos de encefalopatía moderada a grave secundaria a estado postictal.
El cuadro clínico de estado epiléptico asociado a anemia microcítica arregenerativa, trombocitopenia y esplenomegalia gigante puede corresponder a EG tipo III o a talasemia beta leve.
En el servicio de oftalmología y retina se encuentra lo siguiente: agudeza visual (AV) ojo derecho (OD) cuenta dedos (CD) a 2 m y ojo izquierdo (OI) CD a 4 m, con movimientos sacádicos y parálisis de la mirada vertical. No hay alteraciones en los párpados, la conjuntiva o el segmento anterior. Fondo del OD: medios transparentes, imágenes blanquecinas redondeadas flotantes en el vítreo que tienden a confluir formando un anillo; nervio inclinado con imagen que sugiere una hemorragia en el centro; en la retina hay atrofia generalizada del epitelio pigmentario (Figura 1 A). Fondo del OI: medios transparentes, imágenes blanquecinas redondeadas flotantes en el vítreo, con tendencia a confluir en la arcada temporal inferior; nervio óptico inclinado; en la retina hay atrofia generalizada del epitelio pigmentario (Figura 1 B).
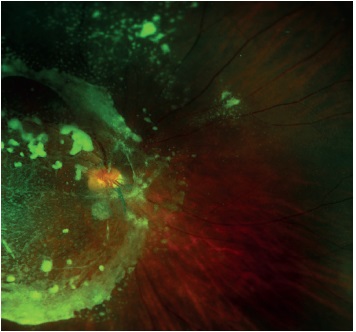
Figura 1 A. Ojo derecho. Imágenes blanquecinas redondeadas flotantes en el vítreo, que confluyen formando un anillo; nervio inclinado con imagen sugestiva de una hemorragia en el centro; atrofia generalizada del epitelio pigmentario de la retina
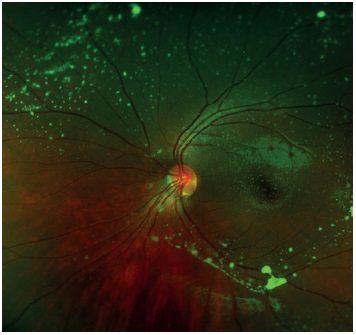
Figura 1 B. Ojo izquierdo. Imágenes blanquecinas redondeadas flotantes en el vítreo, que confluyen en la arcada temporal inferior; atrofia generalizada del epitelio pigmentario de la retina
Angiografía fluoresceínica (OD): hiperfluorescencia peripapilar, lesión hiperfluorescente adyacente a la arcada temporal superior con centro hipofluorescente; en la fóvea se observa hiperfluorescencia de intensidad moderada con patrón petaloide que persiste durante todo el examen y que sugiere edema macular cistoide. Se observa además un patrón de bloqueo secundario a las opacidades vítreas (Figura 2 A). OI: se observan imágenes de bloqueo secundario a las opacidades vítreas sin otras anormalidades (Figura 2 B).
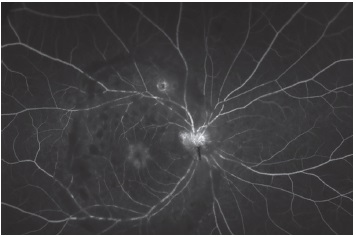
Figura 2 A. Angiografía fluoresceínica del ojo derecho. Hiperfluorescencia peripapilar, lesión hiperfluorescente adyacente a la arcada temporal superior con centro hipofluorescente; en la fóvea se observa hiperfluorescencia de intensidad moderada con patrón petaloide, que sugiere edema macular cistoide. Igualmente se observa un patrón de bloqueo secundario a las opacidades vítreas
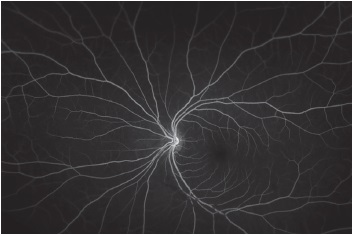
Figura 2 B. Angiografía fluoresceínica del ojo izquierdo. Se observan imágenes de bloqueo secundario a las opacidades vítreas sin otras anormalidades
Ante los hallazgos descritos se hace tomografía de coherencia óptica (OCT, por la sigla en inglés) de ambos ojos. OD: en el vítreo se observa imagen hiperreflectiva que genera efecto de sombra hacia las capas más externas y se correlaciona con los hallazgos anteriores; el vítreo se observa adherido a la retina, hay imagen de fibrosis hacia las capas más internas de la retina que podría corresponderse con las condensaciones en la interfaz vitreorretiniana, se observan rectificación de la fóvea con tendencia al aplanamiento y presencia de quistes en el área foveal. El mapa de grosor muestra un engrosamiento central hasta de 346 µm (Figura 3 A). OI: dentro de límites normales (Figura 3 B).
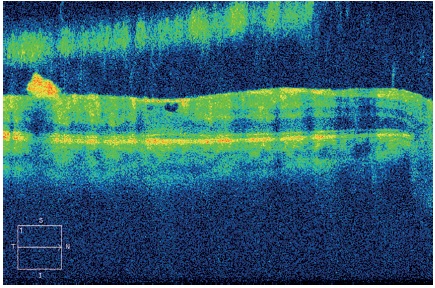
Figura 3 A. Tomografía de coherencia óptica del ojo derecho. Se observa imagen hiperreflectiva en el vítreo que genera efecto de sombra hacia las capas más externas; hay imagen de fibrosis en las capas más internas de la retina que podría corresponderse con las condensaciones en la interfaz vitreorretiniana; rectificación de la fóvea y presencia de quistes. El mapa de grosor muestra engrosamiento central hasta de 346 micrómetros
En el aspirado de médula ósea se observan células de Gaucher.
El cuadro clínico sugiere el diagnóstico de EG tipo III por defecto de betaglucocerebrosidasa. En la clasificación de esta enfermedad, el compromiso neurológico y visceral y la evolución lenta sugieren el tipo 3B. El resultado de la prueba genética confirma el diagnóstico, pues muestra una mutación homocigota en el gen GBA (c.1448T>C p.L483P) (L444P), la LysoGb1aumentada (180,0 ng/mL; valor de referencia menos de 4,8 ng/mL), y la enzima glucocerebrosidasa disminuida (menos de 3,5 µmol/L/h; valor de referencia más de 4,9 µmol/L/h). Se decide no iniciar la terapia enzimática debido al compromiso del SNC y a la escasa difusión de dicha terapia al líquido cefalorraquídeo.
Con respecto al compromiso oftálmico, se opta por el tratamiento médico del edema macular del ojo derecho con AINE tópicos, dadas las condiciones sociales de la paciente, la poca evidencia disponible sobre los posibles tratamientos quirúrgicos y la probable recidiva de sus lesiones sin un tratamiento enzimático. Luego de mejorar su condición sistémica y permanecer estable durante varios días, se decide darla de alta con seguimiento ambulatorio con OCT de mácula en 4 meses. Lamentablemente, la paciente presenta un nuevo episodio de estado convulsivo y muere en su hospital local en enero de 2015.
Se obtuvo el consentimiento informado escrito por parte del adulto responsable de la paciente para la publicación de este caso y de las imágenes que lo acompañan.
DISCUSIÓN
Aunque infrecuentes, las opacidades vítreas en pacientes con EG son una de las manifestaciones oculares más llamativas de la enfermedad1)(2)(3)(4)(5)(6)(7. En una serie de 80 pacientes con EG tipo1 se encontró una incidencia de opacidades vítreas del 3 %7. La glicosilceramida o cerebrósido es el componente principal de estas opacidades. Esta sustancia se deriva de la degradación de la mielina, los leucocitos, eritrocitos y células endoteliales. Aunque algunos sugieren la posible migración a través de la lámina cribosa de células de la oligodendroglia cargadas de mielina, hasta el momento se desconoce el mecanismo exacto del depósito de glicosilceramida en el segmento posterior3.
El gen que codifica para la glucocerebrosidasa está localizado en el cromosoma 1q y se compone de 11 exones y 10 intrones. A pesar de esta detallada identificación genética, las manifestaciones de la EG son muy diversas8. Incluso se han informado pacientes con las mismas mutaciones y portadores de los mismos genotipos con manifestaciones clínicas disímiles. Una de las explicaciones para la gran variación fenotípica de la enfermedad podría ser la presencia de un área homóloga al gen de la glucocerebrosidasa (pseudogén) cuya similitud es del 96 %, en la cual hasta el momento se han identificado más de 200 posibles mutaciones9.
En nuestra paciente se diagnosticó EG tipo 3B con una mutación homocigota en el gen GBA (c.1448T>C p.L483P), descrita también como L444P, y cuya frecuencia entre las distintas mutaciones es de 3 %10; se la ha relacionado en informes anteriores con el tipo de compromiso vítreo descrito; es, pues, un nuevo caso que se correlaciona con esta mutación y, hasta donde sabemos, es el primer caso reportado en Colombia con estas características. Los hallazgos oculares en esta paciente fueron importantes para el diagnóstico etiológico del síndrome convulsivo, que no era claro en un comienzo.
En algunos pacientes se ha hecho vitrectomía vía pars plana, con buenos resultados a corto plazo desde el punto de vista de la agudeza visual, pero no hay documentación de los resultados a largo plazo ni de las recidivas3)(6. Debido a las condiciones sociales de la paciente y a su afectación neurológica de base, se optó por un tratamiento no quirúrgico desde el punto de vista visual.
Este informe contribuye al entendimiento de esta rara y hetereogénea enfermedad.
REVISIÓN DEL TEMA
Historia
El médico francés Phillipe Charles Ernest Gaucher (1854-1918) fue el primero en describir la enfermedad que lleva su nombre (EG)11. En su tesis De l’épithélioma primitif de la râte; hypertrophie idiopathique de la râte sans leucémie publicada en 1882, Gaucher documentó el caso de una mujer joven con hipertrofia idiopática del bazo causada por infiltración en el parénquima de grandes células nucleadas y amorfas. En la descripción del caso el autor en forma acertada correlacionó la progresión de la esplenomegalia con el desarrollo de epistaxis, púrpura, ictericia y caquexia12.
Las bases bioquímicas de esta enfermedad se entendieron 83 años después de la primera descripción13 y solo en los años 80 se detallaron sus bases moleculares14; sin embargo, la enfermedad mantiene su nombre en honor al médico francés que hizo su descripción detallada.
Epidemiología y clasificación
Con el fin de clasificar la EG es fundamental determinar la presencia y progresión del compromiso neurológico, asignando cada caso a uno de los tres grupos existentes. La clasificación, además, permite explicar a las familias aspectos pronósticos como la expectativa de vida y las posibles discapacidades.
Esta es la más común de las enfermedades de depósito lisosomal y puede presentarse en todas las etnias con una frecuencia de 1/40.000-1/50.000 recién nacidos vivos15, pero la incidencia en judíos asquenazis es mucho mayor que la informada en la población general (1/1200 nacidos vivos con EG1)16.
Hasta la fecha se han informado en Colombia 76 pacientes con EG, de los cuales 55 se encuentran registrados. De estos, 29 son adultos (53 %) y 26, niños (47 %). Cincuenta son del tipo I y 5, de los tipos II y III17.
Clínicamente la EG se caracteriza por su gran variabilidad fenotípica, con un espectro de manifestaciones que pueden ir desde la muerte fetal hasta ser asintomática en pacientes ancianos18.
El compromiso en la EG1, también conocida con el nombre de EG no neuropática, se limita principalmente a los sistemas hematopoyético, esquelético y visceral16. Aunque este grupo en algunos casos se denomina EG del adulto, hasta en 30 % de los pacientes el diagnóstico se hace antes de los 10 años de vida19.
Tanto la EG2 como la EG3 se caracterizan por afectar el SNC. La EG2 (neuropática aguda) se manifiesta desde el período prenatal o durante los primeros meses de vida, y su tasa de mortalidad es del 100 % a la edad de 3 años. A diferencia de la EG2, los pacientes con EG3 (neuropática subaguda) suelen tener un curso más prolongado y logran vivir hasta finalizar la adolescencia e incluso pueden alcanzar la adultez temprana16.
Debido a variaciones en su presentación, la EG3 se puede subdividir en 3a, 3b y 3c. En los pacientes con EG3a suelen presentarse compromiso visceral leve y convulsiones mioclónicas progresivas que llevan a la muerte en las dos primeras décadas de la vida. A diferencia del anterior, el tipo 3b se caracteriza por alteraciones viscerales graves y progresión lenta durante 10 a 20 años de las manifestaciones neurológicas20. Los pacientes con el tipo 3c presentan calcificaciones de las válvulas aórtica y mitral, además de las manifestaciones viscerales y neurológicas11.
Fisiopatología
La EG es un trastorno autosómico recesivo caracterizado por deficiencia de la enzima glucocerebrosidasa, también llamada glucosilceramidasa, cuya función principal es convertir el glicolípido glicosilceramida o cerebrósido (componente de la membrana de los eritrocitos) en glucosa y ceramida. Este déficit enzimático lleva a la acumulación lisosomal de glicosilceramida en el sistema fagocítico mononuclear en diferentes partes del cuerpo. El acúmulo de esta sustancia en los macrófagos les produce un aumento marcado de volumen, con desplazamiento periférico del núcleo y aspecto esponjoso del citoplasma (células de Gaucher).
Manifestaciones oculares
Segmento anterior
Se ha informado la aparente asociación entre el desarrollo de pingüéculas y la EG, que se atribuye al depósito de células de Gaucher en este sitio. Sin embargo, estudios histopatológicos han descartado dicha presencia, al encontrar solo elastosis de los tejidos21.
El compromiso corneal en la EG es variable y se correlaciona con el tipo de mutación. En 1998, Guemes y colaboradores22 informaron el caso de un paciente palestino de 18 años, que presentaba desde los 6 años calcificaciones en las válvulas mitral y aórtica. No tenía afectada la agudeza visual, pero presentaba opacidades pequeñas finas, lineales en las córneas de ambos ojos de limbo a limbo, pero más prominentes en la región central, confinadas hacia los dos tercios posteriores del estroma. El estudio enzimático halló una mutación en el gen de la glucocerebrosidasa con un cambio D409H. Se presume que las opacidades corneales resultan de la acumulación de glucocerebrosidasa en los queratocitos. Se diferencia de la mucopolisacaridosis en que no hay compromiso del espesor total del estroma, sino de los dos tercios posteriores con espacios libres entre las opacidades individuales. La combinación del hallazgo de enfermedad valvular con calcificaciones y opacidades corneales es característica de la variante D409H de la EG, también llamada tipo 3c.
En 2013, Geens y colaboradores23 informaron sobre un paciente de 57 años con diagnóstico de EG, sin afectación de órganos o neurológico. El estudio genético mostró las mutaciones F216 y L444P. Tenía agudeza visual de 20/25 (OD) y 20/40 (OI). En la biomicroscopía se observaba opacidad corneal bilateral en tres niveles. Se evidenciaban zonas dispersas de opacidad subepitelial, el estroma con zonas focales de engrosamiento y opacidad, mayor en el tercio posterior, además de pliegues en la membrana de Descemet confinados al tercio superior de la córnea. El grosor corneal central estaba aumentado hasta 1023 µm en el ojo derecho y 856 µm en el ojo izquierdo. El recuento endotelial era más bajo de lo esperado para la edad, 1794 células en el ojo derecho y 1432 en el ojo izquierdo.
La microscopía confocal in vivo mostraba el epitelio relativamente normal aunque con algunas zonas hiperreflectivas blancas redondeadas. La arquitectura del estroma corneal anterior estaba distorsionada, con queratinocitos intercalados con múltiples puntos blancos pequeños. El estroma profundo era de apariencia espiculada, debido a que los queratinocitos son reemplazados por estructuras en forma de barra (rod-like), que también se han descrito en pacientes con otras enfermedades de depósito.
La parte anterior con queratinocitos de morfología anormal intercalados con puntos blancos diminutos se debe a la vacuolización citoplasmática extensa de los pacientes con EG. En la literatura hay pocos casos descritos de EG con anomalías corneales. Los reportes son en su mayoría de la enfermedad con mutación D409H, anteriormente descrita. Los depósitos corneales descritos en la enfermedad F216/ L444P son raros24.
Segmento posterior
Aunque infrecuentes, las opacidades vítreas en pacientes con EG están entre las manifestaciones oculares más llamativas de la enfermedad. En una serie de 80 pacientes con EG1 se encontraron dichas opacidades en 3 %7. Se ha descrito que en estos pacientes es más frecuente este hallazgo después de la esplenectomía debido a más circulación de glucosilceramida3. Shrier y colaboradores3 describieron en una paciente tortuosidad vascular marcada, con un patrón en espiral similar al de la enfermedad de Fabry, al igual que la presencia de membrana epirretiniana.
Sheck y colaboradores25 describieron otro caso de condensaciones vítreas peripapilares y perivasculares en el polo posterior, en un paciente con EG3 con mutación L444P; las estudiaron con OCT y las describieron como lesiones hiporreflectivas prerretinales en la interfaz vitreorretinal asociadas a desprendimientos localizados del vítreo posterior. Las opacidades persistieron a pesar de la terapia de reemplazo enzimático (TRE).
En 2012, Bruscolini y colaboradores26 describieron el caso de una paciente de 27 años con diagnóstico de EG que presentaba en la retina oclusión de la rama arterial ínfero-temporal confirmada por angiografía fluoresceínica. Fue tratada con esteroides orales asociados a TRE y hubo recuperación completa de la visión.
Se ha asociado tanto a los pacientes con EG1 como a los portadores heterocigotos con un riesgo aumentado de padecer enfermedad de Parkinson. El riesgo era mayor en los pacientes con hiposmia, alteración cognitiva y compromiso motor. Además, estos pacientes y los de enfermedad de Parkinson presentan adelgazamiento retiniano. La posible explicación es la pérdida de la función de la glucocerebrosidasa que puede llevar a estrés oxidativo y disfunción mitocondrial en las células ganglionares27.
Seidova y colaboradores28 describieron el caso de una paciente de 23 años, visualmente asintomática (AV sin corrección 20/20 en ambos ojos), cuyo electrorretinograma evidenció una respuesta fotópica normal, con disminución en condiciones escotópicas del 50 % de la onda B y del 40 % en la respuesta general. Dicha alteración en la función retiniana podría explicarse por cambios iniciales en las células de Mueller y de otras células gliales debido a la absorción de glucosilceramida, más que por los depósitos prerretinianos.
Uveítis
La uveítis es otra alteración descrita en la EG. En 2 (0,4 %) de 527 pacientes con EG1 con mutación N370S29, se describió la presencia de uveítis. Uno de ellos presentaba catarata subcapsular posterior asociada a edema macular cistoide, con buena evolución clínica con el uso de esteroides tópicos. El otro paciente presentaba panuveítis bilateral asimétrica, con precipitados queráticos y vitreítis, y no tuvo mejoría con la TRE.
Músculos extraoculares
En las variedades con compromiso neurológico (EG2 y EG3) se han descrito alteraciones en la motilidad ocular. Las mutaciones más frecuentes en esta alteración fueron la D409H y la T1448C. Entre las anormalidades de dicha motilidad se incluye la falla en los movimientos sacádicos horizontales, sacadas horizontales e inferiores lentas, así como un reflejo oculovestibular anormal y alteración del nistagmo optoquinético10)(30)(31.
TRATAMIENTO
Se dispone actualmente de cinco tipos de tratamiento para la EG1, tres de ellos son TRE y dos, terapias de reducción del sustrato (TRS).
Terapia de reemplazo enzimático (TRE)
La TRE actúa supliendo la enzima glucocerebrosidasa defectuosa en los pacientes con EG; estas enzimas activas catalizan la hidrólisis de la glucosilceramida en glucosa y ceramidas, reduciendo su acumulación en diferentes órganos como hígado, bazo y medula ósea32.
La primera TRE se introdujo en 1991; fue la alglucerasa (ceredasa) derivada de los macrófagos placentarios33. Posteriormente, en 1994, la FDA (Food and Drug Administration) aprobó la miglucerasa (cerezime), que es un análogo de la glucocerebrosidasa producido a partir de células ováricas de hámster y que reemplazó a la alglucerasa. En 2010 se aprobó la tercera TRE llamada velaglucerasa alfa (Vpriv®), producida a partir de líneas celulares de fibroblastos humanos. En 2012, la FDA aprobó la taliglucerasa alfa (Elelyso®), que se produce a partir de células vegetales (zanahoria)32.
Se considera que la TRE es el estándar de oro en el tratamiento de la EG1 y de las manifestaciones no neurológicas de la EG3: mejora el nivel de hemoglobina y el recuento plaquetario, y disminuye el dolor óseo y el tamaño hepático y esplénico34)(35. Esto lo demostraron Anderson y colaboradores34 en el estudio de la cohorte NCS-LSD, en el que 24 pacientes recibieron TRE.
No hay evidencia de que la TRE, incluso en dosis altas, pueda prevenir o enlentecer el deterioro neurológico en los pacientes con EG tipos 2 o 3. No se recomienda esta terapia para los pacientes con EG tipo 2, cuyo tratamiento es de soporte por su mal pronóstico35.
La morbilidad y mortalidad limitan el uso del trasplante de células madre hematopoyéticas en pacientes con EG tipo 1. Además, la TRE es muy efectiva. Sin embargo, el trasplante tendría el beneficio teórico de estabilizar los síntomas neurológicos35.
Terapia para reducción del sustrato (TRS)
La TRS actúa en el ciclo de la síntesis de glucosilceramida, reduciendo su flujo al lisosoma. El miglustat (Zavesca®) es un análogo sintético de la D-glucosa, que actúa inhibiendo la enzima glucosilceramida-sintasa, responsable de la síntesis de glucosilceramida y de otros glicoesfingolípidos, por lo que reduce su cantidad y previene el flujo al interior del lisosoma. Fue aprobado en 2002 por la Agencia Europea de Medicamentos (EMA, por la sigla de European Medicines Agency) y en 2003 por la FDA, para el tratamiento de la EG tipo 3. Puede penetrar la barrera hematoencefálica, y es una posible terapia para las formas neuropáticas; sin embargo, los estudios en pacientes con EG tipo 3 no han demostrado mejoría de los síntomas neurológicos36)(37. Además, se ha relacionado con alta incidencia de efectos adversos como temblor, diarrea, pérdida de peso y trombocitopenia38. En el momento solo está indicada para los pacientes que no toleren la TRE debido a reacciones anafilácticas32.
El eliglustat tartrato (Genz-112638), aprobado por la FDA en 2014, es un análogo de las ceramidas que también inhibe la glucosilceramida-sintasa. Se han observado aumentos significativos del recuento plaquetario y el nivel de hemoglobina, además de disminución en el tamaño del hígado y el bazo. Igualmente, disminución de la infiltración de células de Gaucher en la médula ósea. No penetra la barrera hematoencefálica39.
Tratamiento de soporte
Se puede utilizar en combinación con la TRE o de manera aislada. Para el tratamiento del compromiso óseo se usan los bifosfonatos como el alendronato, además de calcio y vitamina D, que mejoran la densidad mineral ósea y disminuyen el riesgo de fracturas40)(41. Sin embargo, no mejora las lesiones focales como las deformidades óseas vertebrales, pélvicas y femorales32.
Para el tratamiento de soporte de la trombocitopenia, se recomienda el acetato de desmopresina (Ddavp®). Las deficiencias de factores de coagulación específicos se pueden corregir utilizando los factores recombinantes; si no se logra detectar el tipo específico, está indicado el uso de plasma fresco congelado42. La esplenectomía está indicada cuando hay trombocitopenia grave u organomegalia sintomática que no responda a la TRE32.
Futuras terapias
Mediante estudios en animales se está investigando el uso de la terapia génica, utilizando virus como vectores para el reemplazo de los genes mutados, con resultados promisorios43)(44.
Tratamiento de las manifestaciones oculares
En dos hermanas con EG con afectación neurológica se probó el miglustat, con mejoría de los movimientos oculares después de dos años de tratamiento. Sin embargo, la evidencia es contradictoria al respecto, porque no se ha podido demostrar la eficacia de este medicamento en el control de los síntomas neurológicos30.
Los cambios en el segmento posterior y los cambios funcionales en la retina de los pacientes con EG generalmente no mejoran con la TRE, aunque avancen más lentamente. Esto probablemente se deba a que estos medicamentos no pasan la barrera hematorretiniana28)(45. Sin embargo, en 1998 se informó el caso de una paciente de 42 años con EG tipo 1 homocigótica para la mutación N370S, quien presentaba disminución de la agudeza visual del campo izquierdo de 9 meses de duración. Al examen físico había signos de uveítis y vitreítis. No respondió al tratamiento con esteroides intravenosos durante 4 semanas, por lo que se le inició TRE con alglucerasa, por 5 meses. Hubo mejoría de la agudeza visual a 20/30 y de los signos de uveítis y vitreítis46.
En 2011, Rapizzi y colaboradores6 reportaron el caso de una paciente de 16 años con EG tipo 3 con mutación homozigota L444P, que estaba en tratamiento con TRE. Su AV era 20/63 (OD) y 20/25 (OI). Al examen físico presentaba opacidades vítreas blanquecinas y densas en ambos ojos, más en el izquierdo. La ecografía y la OCT mostraron, además, desprendimiento del vítreo posterior y membrana epirretiniana macular en ambos ojos. Se le hicieron vitrectomía parcial y remoción de la membrana limitante interna en el OD. No se removió el vítreo periférico. Tres meses después del procedimiento la AV mejor corregida fue de 20/40 (OD) y 20/32 (OI). Doce meses después alcanzó 20/25 en el OD. Dos años después se mantuvo en el OD, pero decayó a 20/100 en el OI, con evidencia de contracción macular. Se hicieron vitrectomía y remoción de la membrana limitante interna. Tres meses después se restauró la anatomía macular y llegó a 20/32. Al final del seguimiento la AV en el OD fue de 20/25 (3 años después de la vitrectomía) y 20/20 en el OI (1 año después de la vitrectomía).
En 2004, Shrier y colaboradores3 informaron un caso similar, también con buenos resultados 6 meses después de la vitrectomía: AV de 20/60. No se conoce la evolución posterior.
PRONÓSTICO
Con base en el estudio posmortem de 20 pacientes mayores con EG tipo 1, se ha propuesto una posible asociación entre la EG y malignidades hematológicas47)(48. El mieloma múltiple ha sido la neoplasia más frecuente (aumento del riesgo en 5,9 veces). Una posible causa sería el estímulo anormal sobre los macrófagos para la acumulación de glucolípidos49.
Los pacientes con EG tipo 1 usualmente presentan durante la adultez hepatomegalia y esplenomegalia con o sin crisis óseas. La TRE es efectiva porque reduce las visceromegalias, aumenta los niveles de hemoglobina y plaquetarios y mejora el pronóstico50.
Como ya se dijo, la EG tipo 2 es rara, neurodegenerativa, lleva a la muerte antes de los dos años de edad, debido a las complicaciones graves en el SNC; la mayoría de estos pacientes mueren de neumonía u otras infecciones51.
La EG tipo 3 es de presentación más tardía y progresión más lenta42. La mayoría de los pacientes llegan a la adultez, en promedio hasta la tercera década de la vida34)(52.

