











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Fragile X Syndrome (FXS, OMIM 300624) is a genetic disease inherited through the X chromosome and falling under the umbrella of the trinucleotide repeat disorders. FXS is caused by changes in the number of CGG repeats, greater than 200, in the 5’UTR of the Fragile X Mental Retardation 1 (FMR1) gene mapping on Xq27.3, producing methylation of this gene and preventing the transcription and consequently the absence of Fragile X Mental Retardation protein (FMRP)1,2.
FXS prevalence has been estimated at one in 5000 males and one in 4000-8000 females3. No epidemiological data are available in Colombia. The FXS phenotype of those affected is variable and is more evident in men after adolescence, who can present with intellectual disability (ID) ranging from mild to severe; physical findings, including long face, large or winged ears, and macroorchidism are often observed4,5. Approximately 25 percent of women with the full mutation have ID with an IQ below 70. However, up to 70 percent may present deficits in learning including executive functions and emotional difficulties6,7 and May, or may not, present typical facial features4. Premutation carriers with repetition of CGG triplets between 55 and 200, are at high risk to develop Fragile X-associated Tremor/Ataxia Syndrome (FXTAS) or Fragile X-associated Primary Ovarian Insufficiency (FXPOI)8,9,10.
The aim of this publication is to report a family affected by fragile X syndrome and contribute to the Colombian epidemiology of this syndrome. Also, we would like to emphasize the importance of identifying and diagnosing affected individuals with a premutation and a full mutation without phenotype for genetic counseling and education on associated pathologies.
MATERIALS AND METHODS
A sample of peripheral blood leukocytes was obtained by pricking a finger and removing 2 drops of blood immediately placed on a special collection paper - 903™ Protein Saver Card (Whatman™), Blood spots card were saved at room temperature from each of the subjects included in the study. In partnership with the MIND Institute at the University of California, Davis, a PCR screening test, using specific primers that amplify the area of the FMR1 gene containing the CGG repeat, was used to detect the presence of an expanded allele. The PCR approach was performed directly on the blood spot cards as previously described11,12.
Signed informed consent forms were obtained from all participants accordingly to the University of California, Davis, Institutional Review Board-approved human subject protocols.
RESULTS
Here we report the study of a family through an index case that was referred to the medical genetics and dysmorphology consultation at Hospital Universitario del Valle for further evaluation due to the presentation of ID of unknown etiology and typical facies. Clinical findings suggested the presence of FXS and a DNA molecular screening test was performed.
The FXS diagnosis was confirmed and cascade testing was performed on all family members who agreed to participate to the study, which included six women and two additional relatives with ID and physical characteristics of FXS.
DNA testing
Using PCR analysis identified four patients with the number of CGG triplets in the premutation range (55- 200 CGG repeats) and two patients with greater than 200 CGG repeats (full mutation)
Proband, III-4
A 19-years-old male who attends a medical genetics consultation at Hospital Universitario del Valle, was referred by a clinical neurologist, with a diagnosis of ID of unclear origin, possibly genetic due to the presence of facial dysmorphism.
Son of a 30-years-old woman, G1P1, with a history of an uncomplicated pregnancy, cesarean delivery for dystocia, birth weight 2500 gr. During childhood fulfilled neurodevelopmental milestones: first words at 7 months, walking at 12 months and sphincter control at 3 years of age. At 4 years old he had a convulsive episode that was not further studied. Started school at 5 years but began having learning problems, exhibiting hyperactivity and poor response obeying orders.
His teachers, the school psychologist and his parents concluded that the child had ID; not using the diagnostic aid of a neuropsychological test. Therefore, his mother decided to place him in a special school starting at second grade.
As part of his medical history he presented divergent strabismus in the left eye that required surgical management. Currently he has partial reading capabilities, can not write, he manages to count but not to add. Attends sports practices for swimming and soccer.
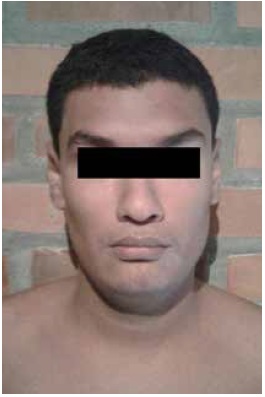
On physical examination height 179 cm, weight 89 kg, BMI 27.8 kg /m2. As particular features he has a long face, large ears and prognathism. Normal hearing screening. He remains attentive and collaborates during the evaluation and shows no aggressiveness. He uses a language with few words, non-repetitive (Figure 1).
Case 2, III-1
Case 2 is a 19-years-old, male, without pre and perinatal complications. The mother affirms that he has ID evidenced by teachers and the psychologist at school; no neuropsychological or IQ testing during childhood.
He is currently enrolled in ninth grade through an inclusion program for academic support. He has had attention deficit hyperactivity disorder (ADHD) since childhood and delay in reaching developmental milestones. Recurrent crying at night, he has exaggerated tantrums, enuresis and inadequate toilet training until the age of 8 years.
Medical history revealed two tonic-clonic seizure episodes, which were handled with valproic acid for a year and recurrent otitis media. Underwent adenoidectomy, hydrocele repair and removal of 17 supernumerary teeth. Currently the patient is behaviorally sociable and not aggressive; however, in particular situations, such as gathering with large groups of strangers and/or loud noises he show high levels of anxiety.
On physical examination height 179 cm, weight 113 kg, BMI 35.29 kg/m2, cephalic perimeter: 60 cm, long face, broad forehead, synophrys, high palate, prognathism, large ears (7.5 cm x 4,5 cm), joint hypermobility (Beighton 5/9), thoracolumbar scoliosis and abdominal striae (Figure 2).

Case 3, II-1
Case 3 is an adult male, 37-years-old, with mild ID and typical facial features. To date he has not had any diagnosis. He is the son of a 42-year-old woman, G5P4, Pregnancy was without any medical complications, with adequate prenatal care, and two ultrasounds reported normal and vaginal delivery at full term pregnancy. At birth, parents were notified of premature closure of the anterior fontanelle, however, in follow up appointments head circumference was reported to be normal. At 8 months he was diagnosed with meningitis, for which he received inpatient antibiotic treatment. He walked at 14 months.
He experienced delay in language: the first syllables were pronounced at 18 months, constructing appropriate sentences at the age of 5 years. At 4 years he required adenoidectomy. Started school at age 7, showing significant learning difficulties, no reported neuropsychological or IQ testing; however, he finished his high school education at the age of 19 years without achieving all the goals. He subsequently pursued studies in culinary. Currently, he has reading and writing capabilities, with difficulty in mathematics. He wears prescription glasses for severe myopia in the left eye and he experiences recurrent conjunctivitis.
On physical examination, his height is 180 cm and weight 113 kg, BMI: 34.8 kg/m2, cephalic perimeter: 61.5 cm. He has long face, large ears (7.7 cm x 4.1 cm), arched palate, dental crowding, prognathism and joint hypermobility (Beighton 5/9). He remains attentive and collaborative during the evaluation and shows no aggressiveness (Figure 3).

Figure 3 Case 3, II-1. A 37-years-old male with full mutation. Physical features include long face, large ears and prognathism
Molecular testing for FXS using blood spot cards showed the presence of a full mutation with an expanded allele of > 200 CGG repeats in two of the cases described above (II-1 and III-1 see Figure 4a and b). The proband was previously tested in Cali, Colombia.
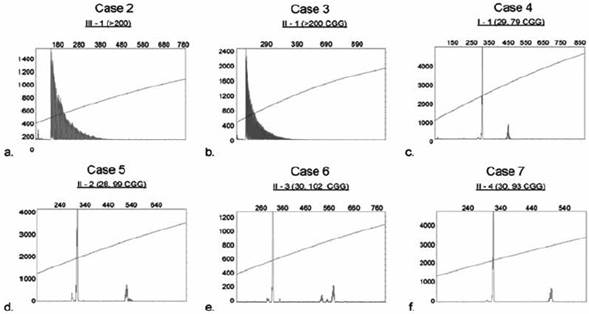
Figure 4 PCR analysis was performed for each case and CGG allele size was determined by capillary electrophoresis (CE) as shown in a-f. The X-axis indicates the size of the alleles in base pairs while the Y-axis indicates the fluorescence intensity of each allele. Plots a and b (Case 2 and Case 3) demonstrate the presence of a full mutation as reflected by the presence of uninterrupted serial peaks13. Plots c-f (Case 4-7) show in all cases the presence of a premutation pattern, with the normal and premutation allele, which sizes are depicted in parenthesis
Cases 4, 5, 6 and 7 - Carriers of premutation allele
Cases I-1 II-2 II-3 and II-4 were four premutation women carrying an allele of 79, 99, 102 and 93 CGG repeat number respectively (Figure 4c-f). These patients of 70, 44, 45 and 50 years of age had no reproductive expectations and no history of ovarian failure before age 40 or difficulty becoming pregnant; nor referred to take medications for anxiety, depression problems or sleep disorders. On physical examination they did not have distal tremor or gait disturbance compatible with ataxia, did not report symptoms of peripheral neuropathy and no positive signs were found on evaluation, therefore it was not considered necessary to perform specialized tests such as electromyography. Furthermore, they did not show cognitive deficit. It was concluded that they did not have clinical signs of FXTAS. No brain images such as Magnetic Resonance (MRI) were available for further evaluation of these patients.
Case 8, II-5
A 51-year-old woman, reached 9th grade in school, could not continue her education because of family problems. She had no children, attempted several medical treatments without achieving pregnancy. She underwent hysterectomy at 40 years of age due to menstrual disorders that caused her excessive vaginal bleeding. Currently she denies experiencing tremor or gait disturbance. She does not take any medication and denies psychiatric problems. DNA molecular testing was not performed because she resides abroad.
The pedigree of the family including the cases identified and carrying the FMR1 mutation is shown in Figure 5.

Figure 5 Pedigree of affected family. This pedigree shows the classic pattern of Fragile X Syndrome, women carrying the premutation had sons with full mutation showing the expansion in the number of the trinucleotide CGG repeats. The squares in the upper left corner denote affected males (full mutation); the central dots represent female carriers (premutation); small centered squares represent unaffected females (normal allele); the small square in the lower right corner indicates an individual that was not tested (NT); open symbols represent normal individuals
DISCUSSION
Fragile X syndrome was first described in 1943 by Martin and Bell, as an X-linked condition that causes inherited ID. In 1991 Verkerk et al. identify the FMR1 gene locus Xq27.3, corresponding to the fragile site sensitive to folate deficiency (FRAXA). Its mutation is described as the cause of fragile X syndrome and it is characterized by the presence of a CGG trinucleotide repeat in the in the promoter region of the FMR1 gene14,15,16. Depending on the degree of the CGG expansion four classes of alleles have been defined: Class I, normal alleles with 5-45 CGG repeats; Class II with 45 to 54 CGG repeats and named intermediate or gray zone alleles; Class III or premutation alleles with 55 to 200 CGG repeats; Class IV or full mutation alleles with greater than 200 repeats17,18,19.
The expansion of triplets with more than 200 CGG repeats which lead to gene methylation and lack of FMRP is considered the cause of FXS. However, the direct causes of gene silencing are epigenetic modifications18, as the methylation of cytokines in the gene promoter and deacetylation of histone tails H3 and H4, among others20 lead to absence of transcription of the gene and consequently the absence of FMRP16.
The classic phenotype of patients with FXS includes physical characteristics such as large and winged ears, with a large and elongated jaw, connective tissue abnormalities including joint hypermobility and macroorchidism4. In addition, patients present a variable degree of ID and behavioral symptoms such as shyness, auto and hetero aggression, poor eye contact, anxiety, attention deficit hyperactivity among others21,22. Women with full mutation, depending on the activation rate which represents the activity of the mutated allele, may present the physical phenotype and severe ID or may not present any suggestive finding4. Carriers of the premutation do not present specific physical characteristics. However, they possess an important risk factor for FRAXopathies, leading to 40 % of men10,23 and 16 % of women to present FXTAS10,24 characterized by distal tremors similar to that of Parkinson’s and an ataxic gait; 20 % of female carriers develop FXPOI10,25 and present an absence of menstruation before 40 years of age, including menstrual cycle disorders and decreased reproduction. Also men, as well as women, may present emotional problems, anxiety and sleep alteration, among other disorders26,27.
All three males affected by FXS included in this report presented the classical physical characteristics of the syndrome: long face, large ears, prognathism and connective tissue abnormalities. Additionally, they presented moderate ID and hyperactivity from childhood. However, they did not undergo neuropsychological or IQ testing, which would have documented the depth of the ID and possibly guided with better learning tools the education they received28.
The four women who had the premutation did not have any suggestive signs or symptoms of FXTAS; however, in FMR1 premutation patients, a contrastenhanced brain MRI should be used to identify imaging findings such as hyperintensity of the middle cerebellar peduncle, hyperintensity of the white matter in different zones of the central nervous system as the pontine nuclei, the insula, the splenius of the corpus callosum and the periventricular region; and generalized severe cerebral atrophy29,30. With these findings, the onset of symptoms can be predicted or a baseline parameter can be anticipated31.
Additionally, none of the female carriers reported absolute criteria of FXPOI32. However, case 8 (II-5), who was the only woman unavailable for DNA testing, reported that she had reproductive difficulties despite having a sexual partner and reproductive desire, a finding also reported in 12.6 % of the female carriers of premutation33. None of the carriers had learning difficulties or emotional problems.
Prevalence of FXS is estimated to be one in every 5,000 men and in one of every 4,000-8,000 women in the United States3. In Colombia, national epidemiologic data does not exist; In 1985 Giraldo et al. from the National Health Institute, performed clinical and cytogenetics studies in Colombia, diagnosing four cases in two out of three of the families studied34. Payan, Saldarriaga et al.35, reported that in Ricaurte, a small population to the north of the Valle del Cauca department exists a prevalence for FXS of one in 38 men and one in 100 women, which exceeds the prevalence reported in literature worldwide by 100 times, also using a karyotype with folate deficiency.
The family reported in this study would be the first to be diagnosed with molecular testing for FXS in Colombia with an origin other than Ricaurte. Also, in Colombia, several families with a confirmed molecular diagnosis belong to the Colombian Association of FXS, which suggests that there exists a significant number of affected persons and carriers throughout the country which need individual attention, genetic counseling and targeted treatment. Given that FXS is the primary cause of hereditary ID, it is possible that many cases are still undiagnosed.
Genetic counseling for male carriers of the premutation consists in informing that the probability of having male carrier sons is 0 % since the X chromosome in males is of maternal origin, but the probability of having female carrier daughters is 100 % and these could have children affected by the syndrome36; information should also be given about the risk of developing FXTAS. There were no men with the premutation in this family.
Women with a premutation or a full mutation have a 50 percent risk of transmitting the affected X chromosome with the FMR1 gene alteration during each pregnancy. Women with a full mutation, with or without a clinical phenotype, have a 50 percent risk of conceiving affected male children and 35 percent risk of conceiving daughters with ID. Premutation carriers have the probability of transmitting an expanded allele depending on the number of triplets; when the range is between 55 and 99 CGG repeats, the probability is between 1.8 and 40 percent; those with more than 100 repeats have a probability of having affected children similar to that of women with full mutation36. The women with the premutation, reported in this family: I-1, II-2 and II-4 carried an expanded allele ranging from 55 and 99 CGG repeats and II-3 had 102 CGG repeats allele; however her only daughter (III-2) had a normal allele. These patients had no reproductive expectations at the time of the diagnosis; none of them presented ovarian failure before the age of 40, distal tremors or gait alterations at the time of the evaluation; each one was given information about the risk of presenting FXTAS in the future. It was important for III-2 and III-3 to obtain a negative result which assured them that they were not carriers of an FMR1 expanded allele.
CONCLUSION
A family with a molecular diagnosis of cases with different allelic variants of the FMR1 gene is reported.
The importance of molecular diagnosis of FXS is presented; not only it is important for the affected patients but for their families, given the implications for healthy people having affected children and developing associated pathologies. Additionally, it contributes to the construction of Colombian epidemiological data on FXS.

