











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La hiperplasia suprarrenal congénita (HSC) hace parte de un grupo de errores metabólicos heredados de forma autosómica recesiva, producto de una mutación genética en una de las enzimas participantes en la síntesis del cortisol. El déficit de cortisol genera una retroalimentación positiva hacia el eje hipotálamohipófisis, estimulando la producción de ACTH y esta a su vez, conlleva a un aumento en el número de células de la corteza adrenal1,2.
La principal causa de HSC se debe a la deficiencia de 21-alfa-hidroxilasa y representa aproximadamente el 95 % de los casos3. En segundo lugar, está la deficiencia de 11-beta-hidroxilasa (11OH) ocasionada por un defecto en la CYP11B1, codificada en el cromosoma 8q21, afecta aproximadamente a uno de cada 200.000 nacidos vivos4. Cuando hay déficit de 11OH se acumula 11-desoxicorticosterona (DOCA) y 11-desoxicortisol. Debido a que el primero es uno de los mineralocorticoides con menor actividad, en altas concentraciones produce hipernatremia, retención hídrica e hipertensión arterial (HTA) con hipopotasemia, además de valores bajos de renina. Adicionalmente, el bloqueo enzimático de 11OH conlleva a exceso de andrógenos y síntesis periférica de testosterona5,6.
La presentación clínica del hiperandrogenismo en pacientes femeninas, es la virilización de sus genitales externos aun en etapa prenatal, mientras que en la población masculina debuta años más tarde como una pubertad precoz periférica7. Aunque la hipertensión se atribuye a un exceso de DOCA, puede aparecer varios años después del nacimiento; incluso de forma paradójica, un recién nacido puede presentar pérdidas de sal y choque hipovolémico8.
PRESENTACIÓN DEL CASO
Paciente escolar masculino de 8 años, quien a sus 2 años de edad presentó aparición de vello púbico y fue evaluado por el servicio de endocrinología pediátrica. El servicio encontró bajos niveles de cortisol y elevadas concentraciones de DOCA, 17-hidroxiprogesterona y 11-desoxicortisol, por lo que se hizo el diagnóstico de HSC por deficiencia de 11OH. A sus cinco años de edad, se hallaron cifras tensionales altas. Fue manejado de forma ambulatoria con prednisolona (5 mg/día), enalapril (20 mg/12h) y nifedipino (10 mg/8h). Debido a sus condiciones sociales difíciles, tiene poca adherencia farmacológica y controles médicos irregulares. Acude al servicio de urgencias por una intoxicación alimentaria de 24 horas de evolución. Nació a término de las 38 semanas de gestación, con peso de 2.800 g y talla de 52 cm, sin complicaciones durante el parto. Aparentemente, no hay consanguinidad de los padres y su talla genética es 168 cm (- 1 DE).
Al examen físico de ingreso al servicio de urgencias pediátricas, se encuentra tensión arterial de 161 /100 mmHg (percentil ≥ 99 más 5 mmHg), frecuencia cardiaca de 88 latidos por minuto, frecuencia respiratoria de 24 por minuto, peso de 33 kg, talla de 139,5 cm (entre +1 y +2 DE), IMC 17,07 (entre 0 y +1 DE). Se observa hiperpigmentación generalizada de areola, genitales, manos, encías y codos (Figura 1). Además, se documenta acné en cara y espalda, soplo cardíaco sistólico grado III/V audible en foco aórtico y abdomen blando con Bloomberg negativo. En genitales presenta vello púbico Tanner 4, pene de 11 cm x 1,5 cm y volumen testicular de 3 mL (Figura 2).
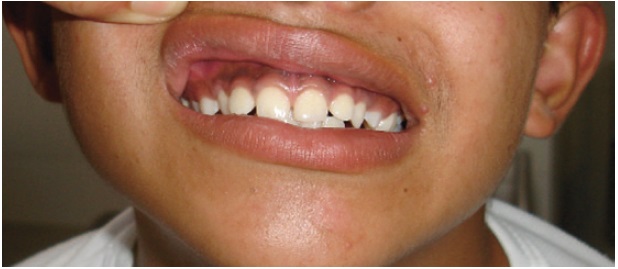
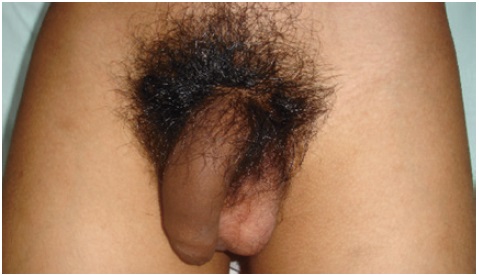
Figura 2 Genitales masculinos externos. Se observa vello púbico Tanner 4, pene de 11 cm x 1,5 cm y volumen testicular de 3 mL
Los laboratorios iniciales muestran un hemograma (Hemoglobina 14,5 g/dL y hematocrito 39,8 %), creatinina (0,65 mg/dL), BUN (17,48 mg/dL), glicemia (72 mg/dL), sodio (137 mmol/L) y uroanálisis normales. Evidencia hipopotasemia (2,6 mmol/L) e hipocloremia (95 mmol/L), cortisol bajo (32 ng/mL con valores de referencia 50-250 ng/mL), renina plasmática baja (4,38 con valores de referencia 20-55 μUI/mL) y un electrocardiograma con hallazgos compatibles con la hipopotasemia (Figura 3). La edad ósea revisada por el método de Greulich y Pyle es de 17 años (Figura 4). En la ecografía renal y de vías urinarias se observa un aumento del tamaño de las glándulas adrenales con diferenciación corticomedular, aspecto más ensanchado de la izquierda, sin evidencia de lesiones focales. El ecocardiograma demostró una cardiomiopatía hipertrófica concéntrica ventricular izquierda no obstructiva, con un aumento de la raíz aórtica e insuficiencia valvular pulmonar leve, sin signos de hipertensión pulmonar. La biomicroscopía del fondo de ojo y agudeza visual se encuentran normales.
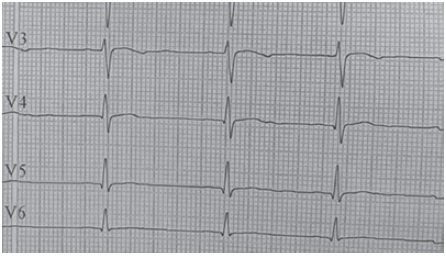
Figura 3 Derivaciones precordiales del electrocardiograma. Onda P aplanada con acortamiento PR. En V3 inversión de la onda T y en V4-V6 aplanamiento de la onda T, hallazgos compatibles con hipopotasemia
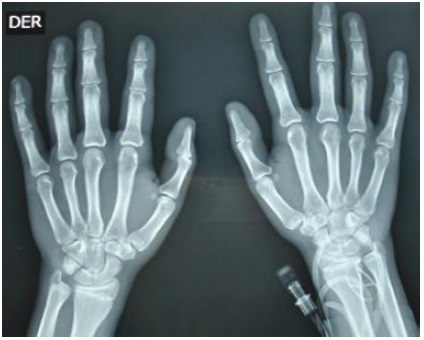
Figura 4 Se observa una edad ósea avanzada por el método de Greulich y Pyle correspondiente a 17 años
Se diagnostica insuficiencia adrenal e hipertensión arterial severa en un paciente con HSC por deficiencia de 11OH mal controlada, se cambia la prednisolona por hidrocortisona a dosis de estrés (100 mg/m2 /día) en infusión continua. Se inicia reposición electrolítica por la hipopotasemia con cloruro de potasio al 4 %. Se evalúa al paciente y se encuentra mejoría de los síntomas iniciales y laboratorios de control que muestran mejoría en los niveles de potasio (3,29 mmol/L) con función renal conservada. Se administra enalapril y nifedipino para el control de las cifras tensionales, sin obtener mejoría. Por esta razón, se escalona y se llega a control de cifras posterior al séptimo día de hospitalización con cinco antihipertensivos: enalapril (20 mg/12h), amlodipino (10 mg/12h), prazosin (1 mg/8h), losartán (50 mg/día) y espironolactona (25 mg/6h). Se intenta retirar gradualmente antihipertensivos para disminuir la polifarmacia, no obstante, debido a la tendencia de cifras tensionales en el límite superior se da egreso con los cinco antihipertensivos. Por la imposibilidad de hidrocortisona oral se maneja ambulatoriamente con prednisolona (5 mg/día) y recomendación de bajo consumo de sal, así como recomendaciones para prevenir la insuficiencia adrenal.
Por la situación geográfica y las condiciones socioeconómicas del paciente, no ha tenido controles regulares y ha requerido el uso de todos los antihipertensivos y la prednisolona oral a igual dosis. No ha vuelto a tener crisis adrenales de acuerdo a la comunicación telefónica con los familiares y está pendiente realizar el estudio de la mutación genética.
DISCUSIÓN
La prevalencia de HTA crónica es de tan solo 1,6 % en la población infantil de los Estados Unidos9 y sus principales causas son los síndromes de exceso de mineralocorticoides4. El exceso de DOCA en la HSC por déficit 11OH genera incremento del volumen plasmático y de la sensibilidad de la musculatura de las arteriolas. Por eso, hasta dos terceras partes de los pacientes, desarrollan HTA crónica infantil y lo hacen usualmente en la etapa de lactante o preescolar10. En el caso presentado al momento del diagnóstico de la HSC no tenía HTA. Tres años más tarde se detectó e inició manejo antihipertensivo. Sin embargo, por el tratamiento inadecuado, se produjo la HTA severa que requirió el uso de cinco antihipertensivos simultáneos para controlar las cifras tensionales. Adicionalmente, como describe Menon et al., la hipertrofia ventricular izquierda dificulta más el manejo de la hipertensión11.
La complicación más frecuente de la HTA crónica no controlada en niños es la hipertrofia ventricular izquierda12. En el caso presentado, se detectaron las alteraciones ecocardiográficas correspondientes a este diagnóstico, similares a las descritas por Ramires et al13. Debido a la severidad y cronicidad de la HTA, era pertinente evaluar la existencia de otras complicaciones. Se realizaron los exámenes necesarios y no se encontraron hallazgos en órgano blanco como retinopatía hipertensiva, falla renal y/o ataque isquémico transitorio descritos en otras series11,14.
La hipopotasemia usualmente tiene manifestaciones clínicas como fatiga, debilidad muscular, parálisis y alteraciones electrocardiográficas15. Se evidenció en el caso presentado una hipopotasemia moderada sin los síntomas anteriormente mencionados, pero sí se observaron los cambios electrocardiográficos, similares a los reportados por Rustagi et al.16. La hipopotasemia se relaciona con la sobre estimulación del receptor de mineralocorticoide, lo cual produce eliminación de potasio en el túbulo contorneado distal y reabsorción de sodio. Cifras tensionales altas y valores bajos de potasio, son individualmente inhibidores de la síntesis de la renina a nivel yuxtaglomerular6, lo que explica la HTA con la actividad de la renina plasmática baja presente en nuestro caso. Por ello se recomienda incluir antihipertensivos ahorradores de potasio como la espirinolactona.
En la síntesis de ACTH se produce paralelamente la hormona estimulante de melanocitos, que en altas concentraciones como ocurre en la HSC, puede ocasionar una pigmentación en piel y mucosas, denominada melanodermia17. Su expresión típica es una hiperpigmentación en pliegues de flexión dorsales de las manos, pies, codos, cicatrices y en las encías16,18, compatible con los hallazgo al examen físico en el caso.
En la pubertad normal, la gonadarquia inicia con el aumento del volumen testicular, estimulado por la secreción GnRH a nivel hipotalámico19. Sin embargo, en el caso, el volumen testicular es prepuber asociado al aumento del tamaño del pene y vello púbico avanzado, lo que es indicativo de una pubertad precoz periférica. El exceso de andrógenos por déficit de 11OH, puede virilizar in útero un feto 46XX y producir un fenotipo con ambigüedad genital; en fetos 46XY, el exceso de androstenediona principalmente conlleva a la pubarquia temprana. El tratamiento irregular de corticoides permitió el progreso rápido de la pubertad precoz. Su crecimiento acelerado es evidente por una talla alta para su talla media parental, característica de la pubertad precoz verdadera. El paciente tenía una edad ósea muy avanzada propia de etapas donde el crecimiento óseo está finalizando, lo que permite inferir la repercusión negativa que este proceso tendrá en la talla final del paciente, como describió Cesario et al.,20.
Se han descrito frecuentes mutaciones del gen que codifica CYP11B1 asociadas la deficiencia de 11OH, especialmente entre judíos marroquíes21. Sin embargo, la mutación R384X hallada recientemente en Colombia por Matallana et al.,22 no había sido descrita. Está pendiente del resultado genético de esta familia, con la posibilidad de hallar un patrón genético propio en la población colombiana.

