











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La candidiasis es una infección oportunista producida en la mayoría de los casos por Candida albicans, un hongo dimórfico que puede hacer parte de la flora normal de la boca, el tracto genitourinario, gastrointestinal, y en menor medida de la piel. Sin embargo, presentaciones como la candidiasis cutánea o mucosa pueden darse en personas sanas, sin necesidad de tener antecedentes de inmunosupresión.
En los niños, particularmente después del período neonatal, estas infecciones a repetición son poco frecuentes, por lo que al presentarse sin haber un factor de riesgo identificado, como historia de uso de antibióticos o esteroides, se debe hacer una búsqueda exhaustiva de comorbilidades, incluyendo las inmunodeficiencias primarias1.
La candidiasis mucocutánea crónica es una manifestación de algunas inmunodeficiencias primarias, que característicamente se presenta en la infancia en el 80 % de los casos de forma heterogénea2,3 con infecciones en la piel, las uñas y la mucosa genital, oral e incluso, oftálmica4. Las cuales pueden ser recurrentes, persistentes o severas5. Estas infecciones recurrentes se desencadenan por múltiples defectos genéticos que conducen a una alteración en la inmunidad innata, principalmente sobre las citocinas del perfil Th17, las cuales son algunas de las interleucinas encargadas de la inmunidad antimicótica1,6. Dependiendo de la mutación genética que se asocie a esta inmunoinsuficiencia, se puede asociar a otras endocrinopatías, trastornos autoinmunes e infecciones no candidiásicas, que en ocasiones pueden ser causantes de gran morbimortalidad7. Es raro que estos pacientes desarrollen este tipo de infecciones de manera sistémica8.
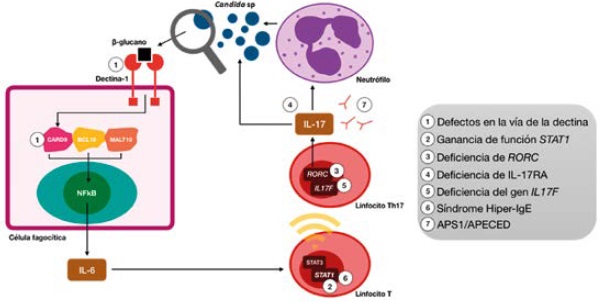
Bajo condiciones normales, las células fagocíticas reconocen, digieren y matan al microorganismo fúngico gracias al reconocimiento del β-glucano de su pared9. Este reconocimiento se logra mediante el receptor tipo dectina-1 de las células fagocíticas, que tiene la capacidad de activar a la proteína 9, miembro de la familia del dominio de reclutamiento de las caspasas (CARD9), que finalmente se une con BCL10 (Bcell Leukemia/Lymphoma 10) y MALT-1 (Mucosa-associated Lymphoid Tissue Lymphoma Translocation Gene 1) para inducir la activación del factor nuclearκβ (NF-κβ) y generar la transcripción de múltiples citocinas proinflamatorias tales como IL-6, IL-23, TNF-α e IL-1β.
La IL-6 induce la transcripción del STAT3 (factor de transductor de señal y activador de la transcripción 3), el cual se une al promotor de IL-17, llevando a los linfocitos T a diferenciarse hacia su perfil Th17 y así lograr la producción de IL-171. Defectos en cualquiera de los pasos mencionados van a llevar a una disfunción en el control de la infección por el hongo (Tabla 1), (Figura 1), lo que hace importante identificarlos ya que se convierten en dianas terapéuticas en este tipo de pacientes10, los cuales históricamente se han manejado de forma tópica con nistatina o sistémica; con fluconazol como primera línea, y en caso de presentarse resistencia (hasta en el 10 % de los casos)11 con itraconazol, voriconazol o posaconazol8,12.
Tabla 1 Inmunodeficiencias primarias causantes de candidiasis mucocutánea crónica
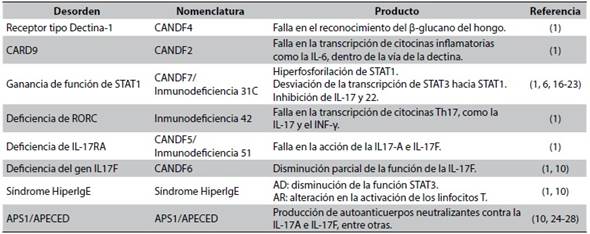
AD: autosómico dominante. AR: autosómico recesivo
Fuente: Elaboración propia
Figura 1. Mecanismos de daño inmunológico en la candidiasis mucocutánea crónica Abreviaturas: CARD9: caspase recruitment domain family member 9, BCL10: B cell leukaemia/lymphoma 10, MALT10: mal regulon transcriptional activator 10, RORC: related orphan receptor C, STAT1: signal transducer and activator of transcription 1, STAT3: signal transducer and activator of transcription 3, NFkB: nuclear factor kappa B , IL-17F: interleukin 17F, APS1/APECED: autoimmune polyendocrinopathy candidiasis ectodermal dystrophy
Los pacientes con poliendocrinopatía autoinmunecandidiasis-distrofia ectodérmica (APECED) se tratan con anfotericina B tópica o sistémica13. Otras terapias que se han planteado en este tipo de pacientes son la infusión de linfocitos donantes, factores estimulantes de colonias de granulocitos, inhibidores de la JANUS kinasa como el ruxolitinib14, inhibidores de la histona deacetilasa y trasplante de médula ósea15.
Defectos en la vía de la dectina
En este dominio pueden presentarse dos alteraciones, una de ellas al existir un defecto en el receptor tipo dectina-1, el cual se va a conocer como candidiasis familiar tipo 4 (CANDF4), y otra respecto al gen que codifica para CARD9, el cual se va a conocer como candidiasis familiar tipo 2 (CANDF2) (Tabla 1). Ambas llevan a la disminución en las células T productoras de IL-17 y por esto a una predisposición a las infecciones, incluso invasivas, por Candida sp; y por otro tipo de feohifomicetos, dermatofitos, Aspergillus sp., Crytococcus neoformans y Pneumocystis jirovecii1.
Ganancia de función de signal Transducer and Activator of Transcription 1 (STAT1)
Su mutación causa un tipo de candidiasis mucocutánea crónica llamada Inmunodeficiencia 31C, antes llamada CANDF7 (Tabla 1). En la última década se han documentado múltiples reportes de esta mutación, la cual se ha encontrado en la mayoría de los casos sindrómicos de candidiasis mucocutánea crónica16. Se cree que esta mutación alélica del gen STAT1 genera una desviación de la transcripción de STAT3, mediada por IL-6, hacia la transcripción de STAT11, de lo que gana función; lo cual también puede ser secundario a una alteración en su desfosforilación17-20, a la vez que genera mejores respuestas a los inhibidores de IL-17, e IL-22, entre otros.
Lo anterior hace que cualquier mutación en este gen lleve, hasta en el 98 % de los casos, al desarrollo de candidiasis mucocutánea crónica en la infancia temprana. Sin embargo, este tipo de pacientes también tienen susceptibilidad a otro tipo de infecciones tales como dermatofitosis6, coccidioidomicosis diseminada21, histoplasmosis, mucormicosis diseminada, estafilocócicas, estreptocócicas, así como también a otras causadas por Pseudomonas sp., Haemophilus influenza, y virus o micobacterias, dependiendo del sitio de la mutación genética, que también puede alterar la producción de INF-γ1,22. En estos pacientes, igualmente se han documentado trastornos autoinmunes como enfermedad tiroidea, vitiligo, diabetes mellitus tipo I y psoriasis; asimismo el aumento de la prevalencia de aneurismas cerebrales y malignidad respecto a la población general1, sobre todo de carcinomas escamocelulares de esófago23.
Deficiencia de Rar-related Orphan Receptor C (RORC)
El gen RORC codifica para RORγ (receptor huérfano gamma relacionado con los retinoides), el cual se expresa en los linfocitos Th17 y funciona como factor de transcripción para la producción de sus citocinas, entre ellas IL-17 e INF-γ (Tabla 1). Es por esto, que mutaciones bialélicas de pérdida de la función de este gen se han documentado en algunos pacientes con candidiasis severa y micobacteriosis, que explicarían la predisposición a estas infecciones1. Estos pacientes también se presentan en algunas ocasiones con hipoplasia tímica y linfopenia leve.
Deficiencia de Interleukin 17 Receptor A (IL-17RA)
La deficiencia del receptor A de IL-17 (IL-17RA) se describió por primera vez en el 2011 de forma autosómica recesiva, llamada en ese entonces CANDF5, ahora Inmunodeficiencia 51 (Tabla 1). Clásicamente se ha descrito que este tipo de pacientes van a tener predisposición para infecciones por Candida albicans y Staphylococcus aureus antes de los 6 meses de vida, pero sin documentación hasta la fecha de otras asociaciones como endocrinopatías, malignidad o aneurismas1. Se plantea que es una deficiencia completa, ya que no permite la función tanto de la IL-17A como de la IL-17F.
Deficiencia del Interleukin 17F (GEN IL17F)
Se identificó por primera vez en una familia en Argentina de forma autosómica dominante, la cual desarrolló candidiasis mucocutánea crónica en el primer año de vida. Se propone que esta deficiencia es parcial, ya que no hay una pérdida completa de la actividad de la IL-17F pero sí hay acción en la IL-17A10, por lo que, en comparación con los pacientes con deficiencia de IL-17RA, van a tener un fenotipo más leve y sin infecciones estafilocócicas1. Esta enfermedad se conoce como CANDF6 (Tabla 1).
Síndrome Hiper-IgE
El síndrome Hiper-IgE es una inmunodeficiencia primaria caracterizada por niveles elevados de IgE sérica, dermatitis, infecciones micóticas o bacterianas, recurrentes en la piel o el tracto respiratorio. Su forma autosómica dominante es causada por una disminución en la función del STAT3, en la que se alteran las señales dependientes de IL-6, entre otras. Lo que finalmente lleva a niveles bajos de IL-17 y a la predisposición a infecciones fúngicas. La candidiasis mucocutánea crónica se ve hasta en el 83 % de estos pacientes, generalmente manifestada como candidiasis oral, genital, cutánea u onicomicosis; una manifestación rara de la enfermedad es con candidiasis invasiva o sistémica. Su forma autosómica recesiva se ha asociado a mutaciones en el gen DOCK8 (Dedicator of Cytokinesis 8) que produce una alteración en la activación de los linfocitos T10, con susceptibilidad mayor a infecciones virales, y en la cual la candidiasis mucocutánea crónica se presenta solo en el 64 % de los casos. De presentarse tiene un compromiso menor (Tabla 1). Otros tipos de síndrome Hiper-IgE no se han asociado con candidiasis mucocutánea crónica1.
APS1/APECED
El síndrome autoinmune de poliendocrinopatía tipo I (APS1), también llamado poliendocrinopatía autoinmune-candidiasis-distrofia ectodérmica (APECED) es un síndrome raro, generado por mutación en el gen regulador autoinmune AIRE (Autoimmune Regulator)24, generalmente autosómico recesivo, que genera producción de autoanticuerpos neutralizantes25 contra múltiples citocinas como la IL-17A e IL-17F en un 41 a 75 % de los casos10, (Tabla 1). Esto explica el desarrollo de la candidiasis25, sin embargo, los niveles de estos anticuerpos no se correlacionan con la gravedad de la misma26. Se ha planteado también, que además de los autoanticuerpos generados contra esta interleucina, AIRE participa en la vía de la dectina-1, por lo que, defectos a nivel genético alteran esta vía predisponiendo en otro nivel a infecciones fúngicas en esta enfermedad27.
Este síndrome se caracteriza por presentar una triada clásica (completa en el 33 % de los pacientes) constituida por candidiasis mucocutánea crónica, hipoparatiroidismo (el cual puede ser la primera manifestación endocrina de la enfermedad28) y la enfermedad de Addison. Sin embargo, pueden presentarse otras manifestaciones endocrinológicas como hipogonadismo hipergonadotrópico, disfunción hipofisiaria, tiroiditis autoinmune, hepatitis crónica, anemia perniciosa28, así como manifestaciones ectodérmicas como alopecia areata, vitiligo, distrofia ungular o dental1. Es raro, pero estos pacientes pueden presentar síntomas respiratorios, polineuropatía y nefritis intersticial tubular28, lo que hace que su diagnóstico y tratamiento sea todo un reto para el especialista.
OTRAS DEFICIENCIAS
El desarrollo de la candidiasis mucocutánea crónica se ha asociado también con deficiencias de IL-17RC (llamada CANDF9) y del TRAF3IP2 (proteína 2 que interactúa con el TRAF3). Esta última proteína, también llamada ACT1 es una molécula que activa, entre otras, la vía del NF-κβ, por lo que al estar mutada genera una abolición completa de la liberación de IL-17A, IL17E e IL-17F desde los fibroblastos y leucocitos, generando una inmunidad fúngica alterada1.
Otro tipo de candidiasis familiares son la tipo CANDF3 y CANDF1, las cuales típicamente solo afectan las uñas y se asocian a una deficiencia de la molécula de adhesión intracelular ICAM-1, sin embargo, hasta ahora no se ha identificado el gen mutante.
Se han documentado otras causas adicionales de candidiasis mucocutánea crónica en los humanos, todas relacionadas a la vía del Th17, como defectos en la activación del NF-κβ, síndrome de DiGeorge e inmunodeficiencia combinada severa, entre otros. Las cuales pueden caracterizarse por susceptibilidad aumentada a infecciones por Candida sp.10, así como también se han reportado algunos casos en pacientes con deficiencias humorales de IgG e IgAfigu29.
CONCLUSIÓN
La candidiasis en la infancia puede ser secundaria a tratamientos antibióticos o inmunosupresores. aunque, en ausencia de estos antecedentes, de la presencia de comorbilidades (tales como diabetes mellitus y VIH) y ante infecciones recurrentes, persistentes y/o severas, se debe sospechar candidiasis mucocutánea crónica, la cual no solo es secundaria a una alteración en la inmunidad mediada por IL-17, sino que va a tener múltiples etiologías genéticas que hacen necesario buscar sus asociaciones sindrómicas con un diagnóstico genómico, para así entender mejor la enfermedad y dilucidar las nuevas oportunidades terapéuticas, posibles.

