











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Alport es un trastorno genético raro caracterizado por mutaciones en los genes COL4A3, COL4A4 o COL4A5, que causa síntesis defectuosa del colágeno tipo IV de las membranas basales 1. Clínicamente se caracteriza por nefritis hereditaria progresiva con hematuria y falla renal, hipoacusia neurosensorial y anomalías oculares 2-4. Este síndrome afecta uno de cada 10.000 individuos 5. Se estima que en Estados Unidos hay aproximadamente de 30.000 a 60.000 personas afectadas 6. No existe información epidemiológica en Colombia 7.
Aproximadamente el 80 % de los casos de síndrome de Alport tienen herencia ligada al cromosoma X, con mutaciones en COL4A5 que codifica para la cadena a5 del colágeno tipo IV, los hombres están más severamente afectados que las mujeres 8-10. El 15 % de casos son autosómicos recesivos con mutaciones homocigotas de COL4A3 o COL4A4, que codifican para las cadenas a3 y a4 del colágeno tipo IV, respectivamente. Están hombres y mujeres afectados con igual frecuencia y severidad 11,12.
La forma autosómica dominante ocurre en el 5 % de los casos por mutaciones heterocigotas de COL4A3 o COL4A4 13, es más leve que las formas autosómicas recesivas o las ligadas al X, presenta una progresión más lenta hacia el estadio terminal de insuficiencia renal 8,14.
Las tres estructuras oculares más afectadas en el síndrome de Alport son la córnea, el cristalino y la retina 3,4.
En la córnea se pueden presentar erosiones corneales recurrentes con dolor ocular agudo, fotofobia, visión borrosa y lagrimeo. El cuadro clínico dura usualmente de dos a cinco días y suele ser recurrente. Se han identificado factores precipitantes como tareas de alta exigencia visual, uso de pantallas de computador y uso de lentes de contacto 2,15. Se puede encontrar también opacidad corneal por anormalidad en la membrana de Bowman 16. Otra enfermedad es la distrofia corneal polimorfa posterior causada por anomalías en la membrana de Descemet, puede ser asintomática o presentarse como una sensación de cuerpo extraño con lagrimeo y fotofobia 17,18.
En el cristalino se puede encontrar lenticono, anterior o posterior, está presente en el 50 % de hombres, pero no en mujeres con síndrome de Alport ligado al X. Sin embargo, si se hereda con carácter autosómico es igual en hombres y mujeres 2. La ausencia de colágeno tipo IV en la cápsula del cristalino hace que esta sea muy frágil, extremadamente delgada y que presente rupturas espontáneas con formación de catarata 1,19, con o sin liberación de proteínas del cristalino a la cámara anterior 3,20. Si hay liberación de proteínas del cristalino hacia la cámara anterior puede desarrollarse uveítis facolítica, una reacción ocular inflamatoria, en la cual los macrófagos que han fagocitado estas proteínas se ingurgitan y se depositan en la malla trabecular, bloqueando el flujo del humor acuoso, con el consiguiente aumento de la presión intraocular (PIO) 21.
Cuando hay formación de catarata, la extracción de esta en la cirugía es difícil debido al aumento de la elasticidad y fragilidad de la cápsula, con formación de patrón de rueda dentada 22-24.
Dentro de las alteraciones de la retina se encuentra la retinopatía periférica con puntos coalescentes, siendo esta la anomalía retinal más común. Se puede encontrar, además, alteraciones de la pigmentación foveolar, adelgazamiento retinal temporal 25,26, maculopatía en ojo de buey que no afecta la foveola ni mácula central, defectos en el epitelio pigmentario de la retina y en la membrana de Bruch subyacente 27,28, agujero macular lamelar o gigante 29,30, retinosquisis y anomalías de la membrana limitante interna 31,32. Las anomalías de la retina y el crista- lino son las más comunes 24.
Es importante conocer las complicaciones del síndrome de Alport, en especial las del cristalino, ya que la uveítis facolítica asociada a ruptura espontánea de la cápsula anterior puede producir glaucoma secundario al aumento de la PIO, con pérdida irreversible de la agudeza visual (AV), por lo tanto, el propósito de este artículo es brindar información sobre la uveítis asociada al síndrome de Alport y sus secuelas, para ser detectada y tratada oportunamente.
DESCRIPCIÓN DEL CASO
Paciente masculino de 24 años, se presentó por un cuadro de un día de disminución súbita de AV en el ojo izquierdo (OI), asociado a dolor ocular, fotofobia, lagrimeo y ojo rojo. El paciente no presentó antecedentes de trauma ocular, reciente o pasado, ni otras lesiones oculares. En la anamnesis se encontró que aproximadamente seis meses antes inició con disminución progresiva de la AV por ambos ojos no asociada a otros síntomas oculares. Entre sus antecedentes destacó historia personal de varios años de hipoacusia bilateral progresiva con uso de audífono, niveles elevados de creatinina y siete familiares por línea ma- terna con las manifestaciones típicas de síndrome de Alport, dos de ellos (primo y tía) requirieron diálisis por falla renal (Figura 1).
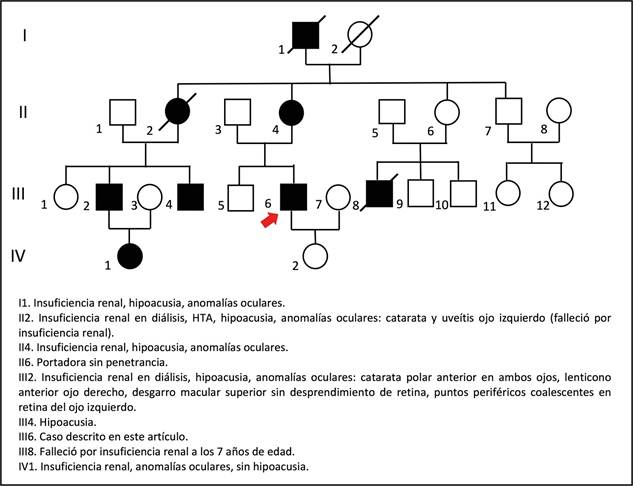
Figura 1 Heredograma compatible con herencia dominante ligada al X. En la descendencia de la pareja I1 - I2 (hombre afectado - mujer sana) no hay transmisión varón-varón, todas las hijas son portadoras del defecto genético: II2 y II4 están afectadas, II6 no está afectada, pero tiene un hijo con la enfermedad, por lo que se asume que es una portadora sin penetrancia. La flecha señala la ubicación del paciente reportado en este artículo (III6). Fuente: elaboración propia.
En la evaluación oftalmológica, se encontró que la mejor AV lejana alcanzada a través de agujero estenopeico fue de 20/30 en el ojo derecho (OD), medida con la cartilla de Snellen y movimiento de manos en OI con reflejos pupilares normales. En la biomicroscopía del OD se evidenció catarata polar anterior central de dos por dos milímetros sin más alteraciones (Figura 2a y 2b). En el OI se observó inyección y hemorragia subconjuntival, catarata con ruptura de cápsula anterior, Tyndall, células 3+ y abundantes restos de material nuclear y cortical del cristalino en la cámara anterior (Figura 2c y 2d). La presión intraocular (PIO) inicial de ambos ojos fue normal, 14 mmHg en OD y 17 mmHg en OI. El fondo de ojo fue normal en OD (Figura 2e) y no valorable en OI. Se realizó ecografía ocular izquierda que mostró el cristalino con un aumento de la densidad, sin otras alteraciones.
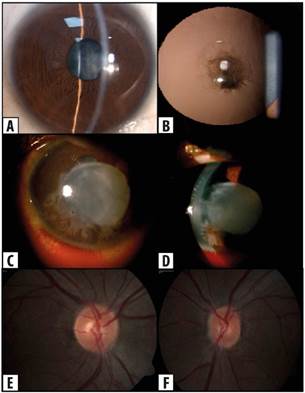
Figura 2 Hallazgos oftalmológicos en paciente con síndrome de Alport. a y b: catarata polar anterior central de dos por dos milímetros en ojo derecho. c y d: Inyección, hemorragia subconjuntival, catarata con ruptura de cápsula anterior y abundantes restos de material nuclear y cortical del cristalino en la cámara anterior del ojo izquierdo. e y f: fondo de ojo normal en ambos ojos, e: ojo derecho y f: ojo izquierdo. Fuente: fotos propias
Se realizaron otros paraclínicos, encontrando creatinina en 5,32 mg/dl, nitrógeno ureico en 47,55 mg/dl, uroanálisis con microhematuria, glucosa 500 mg/dl, proteínas 359,9 mg/dl y cilindros hialinos. En el hemograma se observó anemia leve con hemoglobina en 11,09 g/dl, hematocrito 31,17 %, sin hipocromía ni anisocitosis.
Basados en los hallazgos oftalmológicos observados en la lámpara de hendidura y en los antecedentes personales y familiares del paciente, se realizó el diagnóstico clínico de síndrome de Alport.
Se inició tratamiento tópico con prednisolona al 1 %, una gota cada dos horas y atropina al 1 % una gota cada ocho horas en OI. Al segundo día se encontró que la PIO del OI había aumentado a 29 mmHg, por lo cual se agregó brimonidina 0,2 % más timolol 0,5 %, una gota cada doce horas. Se obtuvo un descenso de la PIO a 14 mmHg. El paciente fue intervenido quirúrgicamente para facoemulsificación y aspiración de restos de cristalino, dejando el saco capsular intacto. La inserción de lente intraocular se programó para un segundo tiempo quirúrgico luego del control de la uveítis. Posterior a la cirugía, se valoró el fondo de ojo izquierdo, el cual fue normal (Figura 2f).
DISCUSIÓN
El diagnóstico de síndrome de Alport dominante, ligado al X en este caso, se realizó clínicamente. El paciente aquí reportado presentó hallazgos en los sistemas auditivo, ocular y renal, es decir, en tres de los cuatro sistemas comprometidos en esta entidad (auditivo, ocular, renal y cardiovascular) 33. Sin embargo, un primo afectado con mayor expresividad sí presentó compromiso en los cuatro sistemas (Tabla 1). Además, los antecedentes familiares del paciente soportan el diagnóstico, ya que tiene siete familiares afectados con fenotipos similares y el heredograma es compatible con un tipo de herencia dominante ligada al X, dado que al analizar la descendencia de la pareja I1 - I2 (hombre afectado - mujer sana) no hay transmisión varón-varón y se encuentra que todas las hijas son portadoras del defecto genético: II2 y II4 están afectadas, II6 no lo está, pero tiene un hijo con la enfermedad, por lo que se debe asumir que ella es una portadora sin penetrancia (Figura 1). De hecho, se sabe que el síndrome de Alport ligado al X tiene una penetrancia incompleta de 95 % 34.
Tabla 1 Manifestaciones clínicas principales y expresividad variable del síndrome de Alport
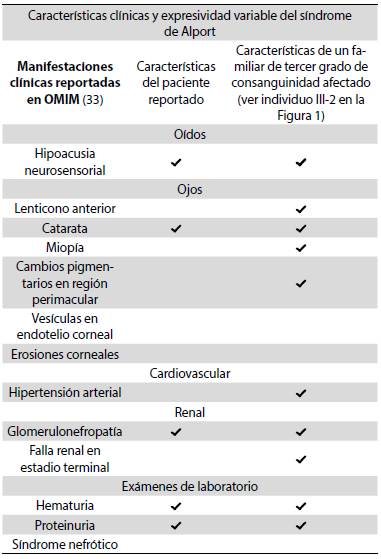
El paciente del caso presenta tres de los cuatro sistemas comprometidos (auditivo, ocular, renal y cardiovascular), sin embargo, un primo afectado con mayor expresividad presenta compromiso en los cuatro sistemas.
El fenotipo ocular del paciente aquí reportado es interesante, dado que se han descrito pocos casos de uveítis facolítica asociada a la ruptura espontánea de la cápsula anterior, incluso en el síndrome de Alport. Se reportó el caso de un paciente masculino de 16 años con síndrome de Alport con ruptura espontánea de cápsula anterior asociada a uveítis facolítica, AV del ojo afectado en percepción de luz, PIO en 49 mmHg y catarata polar anterior en el ojo contralateral, un caso similar al paciente reportado en este artículo 35. Otros casos descritos en la literatura reportan pacientes adolescentes con síndrome de Alport con ruptura espontánea de la cápsula anterior del cristalino, liberación de corteza hacia la cámara anterior, pero sin reacción inflamatoria y PIO normal 36,37. En un reporte de serie de casos de pacientes con síndrome de Alport se encontró que tres de los ocho pacientes descritos presentaron ruptura espontánea de la cápsula y solo uno de ellos, de 4 años de edad, presentó uveítis severa en la cámara anterior por un largo periodo después del postoperatorio de cirugía de catarata, como consecuencia de la presencia de restos del cristalino en la cámara anterior. Los otros dos pacientes tenían 14 y 18 años 38.
La ruptura espontánea de la cápsula anterior puede causar complicaciones graves, incluyendo uveítis facolítica, glaucoma facomórfico y facolítico, que pueden llevar a la ceguera 36. La obstrucción en la red trabecular con proteínas de alto peso molecular, provenientes del cristalino con macrófagos que han fagocitado estas proteínas, causa elevación de la PIO que conduce al desarrollo de glaucoma facolítico 39. La uveítis facolítica es un hallazgo raro que ocurre porque en el síndrome de Alport el grosor de la cápsula anterior es un tercio de lo normal, sufriendo debilitamiento cuando la curvatura del cristalino se vuelve más convexa en el centro durante la acomodación 20,40. Si estas entidades no se tratan adecuadamente pueden producir glaucoma y pérdida irreversible de AV.
No es usual que ocurra ruptura espontánea de la cápsula anterior en la población general 36,37, por lo tanto, si se presenta un paciente con este hallazgo, se debe sospechar de trastornos subyacentes que aumenten la susceptibilidad a sufrirlo. Es importante, ante pacientes con hallazgos fenotípicos infrecuentes, realizar una evaluación clínica detallada en busca de otras manifestaciones clínicas sistémicas y antecedentes familiares que puedan orientar al diagnóstico específico de enfermedades huérfanas, como en este caso.
El síndrome de Alport debe ser diferenciado de otras causas de microhematuria persistente, principalmente el síndrome de Fechtner/Epstein que se caracteriza por nefritis hereditaria, sordera neurosensorial, cataratas, trombocitopenia y macroplaquetas de herencia autosómica dominante 41. Otros diagnósticos diferenciales son la nefropatía familiar IgA, glomerulonefritis membranoproliferativa, nefritis lúpica, glomerulonefritis post infecciosa y nefritis por Henoch- Schonlein 42.
CONCLUSIONES
El síndrome de Alport es una entidad poco conocida, por lo que es importante difundir sus características entre los médicos y especialistas que, de acuerdo con las manifestaciones fenotípicas del síndrome, puedan estar involucrados en su atención, mejorando así la oportunidad del diagnóstico y el tratamiento de los pacientes afectados. El oftalmólogo debe sospechar del síndrome de Alport en pacientes con lenticono, con retinopatía periférica con puntos coalescentes y con ruptura espontánea de la cápsula anterior asociada a catarata.

