











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
En los pacientes con neumopatía crónica, sin origen claro, se debe sospechar aspiración crónica silente a la vía aérea, lo cual es una complicación de muchas enfermedades neuromusculares como las miopatías 1-5.
Las miopatías mitocondriales son entidades poco frecuentes, con una prevalencia aproximada de 1 en 10.000 individuos vivos. Se presentan en cualquier edad, aunque los fenotipos más graves se expresan en los primeros años de vida 6. Las enfermedades mitocondriales más conocidas son el síndrome de KearnsSayre (SKS, por sus siglas en inglés), la oftalmoplejía externa crónica progresiva (por sus siglas en inglés, CPEO), la epilepsia mioclónica con fibras rojas rasgadas (por sus siglas en inglés, MERRF), la miopatía mitocondrial, y la combinación de encefalopatía, acidosis láctica y accidente cerebrovascular (por sus siglas en inglés, MELAS) 7.
REPORTE DE CASO
Niño con 7 años de edad, hospitalizado por cuadro clínico caracterizado por astenia, adinamia, hiporexia, fiebre y dificultad respiratoria. La radiografía del tórax mostró la pérdida de volumen del pulmón izquierdo con opacidad basal ipsilateral (Figura 1).
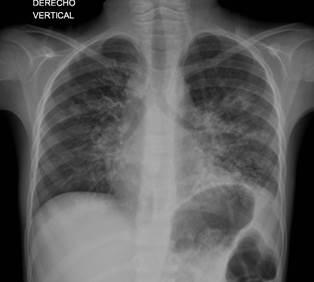
Figura 1 Radiografía de tórax. Evidencia el atrapamiento del aire con pérdida del volumen del pulmón izquierdo, opacidad basal izquierda e imágenes compatibles con bronquiectasias en ambos campos pulmonares. Fuente: propia, con autorización de los padres del paciente para su publicación.
Tenía desnutrición aguda grave (Índice masa corporal/edad -3,35 DE, Talla/Edad -0,52 DE) y evitaba las actividades que requerían esfuerzo.
Sin antecedentes perinatales de relevancia, presentó hipotonía del lactante y un posible trastorno de la deglución. Había tenido múltiples consultas al servicio de urgencias por reagudizaciones respiratorias y un ingreso en la unidad de cuidados intensivos pediátricos por neumonía.
No había consanguinidad entre los padres. El padre tenía antecedente de disfagia y requirió alimentación por sonda en la infancia, posteriormente, se modificó la consistencia de la dieta a una de tipo espeso. Además, presentó debilidad muscular oculofacial faríngea y disfonía, la cual persiste en la actualidad (Figura 2).
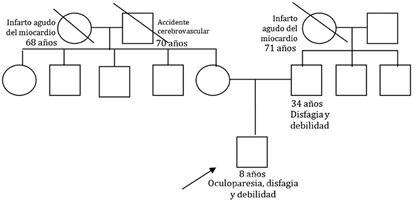
Figura 2 Heredograma. Muestra relación familiar en el mecanismo de transmisión genética de la enfermedad. Fuente: elaboración propia.
Al momento del examen físico se encontró un niño enflaquecido, con disminución del diámetro anteroposterior del tórax, crépitos finos, sibilancias y roncus difusos en ambos campos pulmonares, con acropaquia incipiente. Por la sospecha de neumopatía crónica se solicitó tomografía de tórax de alta resolución, que mostró la pérdida de volumen del pulmón izquierdo y múltiples bronquiectasias (Figura 3), espirometría pre y posbroncodilatador con alteración restrictiva moderada. La ecocardiografía, estudios para tuberculosis, fibrosis quística e inmunodeficiencias primarias y secundarias fueron negativos. La cine radiografía de la deglución reveló el trastorno de la deglución severo en la fase faríngea y los episodios de bronco aspiración silente.
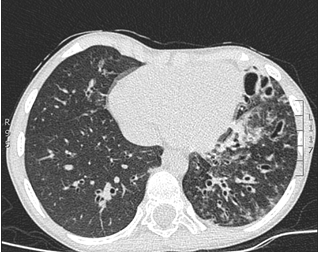
Figura 3 Tomografía de alta resolución de tórax. Se observa la presencia de cambios fibróticos subpleurales apicales bilaterales de predominio derecho, patrón en vidrio esmerilado hacia regiones parahiliares bilaterales de predominio izquierdo, múltiples bronquiectasias, atrapamiento del aire y pérdida de volumen del pulmón izquierdo. Fuente: propia, con autorización de los padres del paciente para su publicación.
El examen neurológico encontró oculoparesia externa multiplanar, especialmente para la supraversión vertical, disminución de los movimientos sacádicos, presencia de reflejo optocinético, paresia del orbicular ocular y labial en forma bilateral sin paresia lingual, con disfonía y voz nasalizada, hipotrofia e hipotonía muscular de distribución axial y cefaloparesia leve con incapacidad para tocarse el pecho con el mentón. Se encontró hiporrefléxico, con fuerza 4/5 para flexión de hombros, flexión de cadera y extensión de la rodilla (según la Escala de fuerza muscular modificada del Medical Research Council (MRC) y el Signo de Gowers negativo.
Se sospechó miopatía congénita hereditaria, tipo enfermedad mitocondrial con oftalmoplejía crónica, síndrome miasténico congénito o enfermedad de Niemann-Pick tipo C. La electromiografía confirmó la enfermedad intrínseca de la fibra muscular en grupos musculares axiales. Se descartó patología de la unión neuromuscular mediante la prueba de estimulación repetitiva de 2 a 20 Hz y la enfermedad de Niemann-Pick tipo C mediante la determinación del valor normal del biomarcador lyso-SM-509. Se solicitó la secuenciación Sanger del gen POLG que mostró la variante alélica patogénica p.Ala467Thr, en homocigosis, lo que confirma miopatía mitocondrial con oftalmoplejía externa crónica progresiva.
Dada la gravedad del trastorno de la deglución, la extensión, irreversibilidad y progresión del daño pulmonar, se realizó una gastrostomía endoscópica percutánea. Además, se formuló coenzima Q10, creatina monohidrato, terapias de fortalecimiento de la musculatura, técnicas de alimentación boca para consistencias seguras y observación expectante de la progresión del trastorno de deglución.
DISCUSIÓN
El proceso de la deglución depende de muchos músculos y acciones neurales reflejas. Se inicia voluntariamente, pero a partir de la fase faríngea continúa bajo el control involuntario 8.
El síndrome aspirativo es la principal causa de la enfermedad pulmonar crónica en pacientes con la enfermedad neuromuscular, causando daño estructural del parénquima pulmonar, enfermedad de la vía aérea y bronquiectasias 4, hallazgos presentes en el paciente. Las aspiraciones repetidas producen alteración del reflejo protector de la tos por la sobreestimulación de los receptores, tornándose silentes. Este paciente ingresó por un problema pulmonar agudo con estigmas físicos y radiológicos de enfermedad pulmonar crónica. Estos hallazgos, aunados a que su padre presentaba características clínicas similares, generaron la sospecha de una enfermedad hereditaria.
Se estableció que el paciente tenía miopatía mitocondrial, un grupo heterogéneo de enfermedades que se presentan por alteraciones genéticas, estructurales y bioquímicas mitocondriales.
Debido a que las mitocondrias son la principal fuente de producción de energía, las características clínicas involucran, típicamente, los tejidos con altos requerimientos energéticos, como son el sistema nervioso, el sistema endocrino y los músculos, particularmente los extraoculares, deglutorios y cardíacos 6.
Estas enfermedades son producto de una alteración en la fosforilación oxidativa en la cadena respiratoria que está controlada por el ADN nuclear (ADNn) y mitocondrial (ADNmt). Los defectos mendelianos mitocondriales pueden abarcar subunidades de los complejos de la cadena respiratoria, las proteínas de ensamble mitocondrial, la traducción del ADNmt, composición lipídica de la membrana mitocondrial interna o la dinámica de la misma organela 9. Solo en el 40 % de los adultos y el 10 % de los niños se encuentra alguna de las mutaciones reportadas 6, en nuestro paciente se identificó dicha mutación.
El gen POLG está ubicado en el locus 15q26.1. El gen se encarga de activar la subunidad alfa de la proteína polimerasa gamma (pol γ), que es la única DNA polimerasa mitocondrial. Este gen tiene un papel crítico en la reparación y replicación del DNA mitocondrial (mtDNA). Las enfermedades relacionadas con los cambios genéticos patogénicos incluyen el espectro de ataxia neuropatía, miocerebrohepatopatía de la niñez, epilepsia mioclónica con miopatía y ataxia sensitiva, oftalmoplejía externa progresiva y síndrome de Leigh. Se han reportado, al menos, 67 mutaciones en POLG relacionadas con CPEO, la mayoría corresponden con cambios en un aminoácido en la subunidad alfa de la POLG, que se traduce en una disminución de la eficiencia para replicar el mtDNA. El síndrome de Alpers-Huttenlocher y la CPEO, con frecuencia, son ocasionados por la mutación POLG Ala467Thr. En la actualidad se habla del término «desórdenes relacionados con POLG» 10.
En la miopatía mitocondrial se afecta gravemente la estructura y función del músculo esquelético por el malfuncionamiento de la fosforilación oxidativa 7,11.
Las fibras musculares tipo 1 se caracterizan por la resistencia a la fatiga y poca fuerza. También son las más afectadas, debido a que contienen abundante cantidad de mitocondrias. Las fibras tipo 2 se afectan en menor medida. En nuestro paciente no se realizó biopsia muscular porque el diagnóstico pudo establecerse a partir de la presunción electroclínica y se confirmó mediante la secuenciación del gen POLG. Actualmente, la biopsia muscular se ha reservado para los pacientes cuyo diagnóstico electroclínico y molecular no es concluyente; en estos casos es fundamental hacer tinción con tricrómico de Gomori, citocromo oxidasa, succinato deshidrogenasa, NADH y microscopía electrónica en el espécimen de patología.
Principalmente, por los hallazgos oculares, el fenotipo del paciente fue específicamente sugestivo de oftalmoplejía externa progresiva crónica (Tabla 1).
La CPEO se caracteriza por ptosis palpebral, alteración de los movimientos oculares con pupilas normales y, en algunos casos, cursa con afectación de la musculatura facial (12,13. Puede asociarse con un compromiso multisistémico con la afectación de la retina, el cerebelo, el corazón y los músculos laríngeos 13-15. Nuestro paciente presentaba trastorno grave de la deglución con aspiraciones silentes y la alteración de la voz, consistente en baja tonalidad y disfonía. Además, presentaba oculoparesia externa multiplanar.
El compromiso respiratorio incluye disminución de la fuerza de los músculos inspiratorios y espiratorios, evidenciado en la disminución de la capacidad vital forzada (CVF), el volumen espiratorio forzado en un segundo (VEF1) y el flujo pico espiratorio (PEF). La debilidad de los músculos respiratorios se correlaciona con la debilidad de la cintura, más no con otras características clínicas como la duración o gravedad de la enfermedad o el tipo de mutación. Estos pacientes son particularmente susceptibles a tener neumonía, dada la presencia concurrente de debilidad laríngea, tos inefectiva por cierre incompleto de la glotis, alteración del tono y la presión del esfínter esofágico superior y alteración del tránsito faríngeo y esofágico, todo lo cual conduce a fenómenos aspirativos. Los trastornos de la deglución se presentan en el 50 % de los pacientes con CPEO 7,14. La neumonía es la principal causa de muerte y puede tener un curso más grave que conlleva a la falla ventilatoria y dificultades en el retiro del soporte ventilatorio. Adicionalmente, los pacientes pueden presentar dificultades en el control de las infecciones 14.
Tabla 1 Características clínicas de la oftalmoplejía externa progresiva crónica por mutación de POL G
| Características clínicas de la oftalmoplejía externa progresiva crónica por mutación POLG (15) | Encontrado en el paciente |
|---|---|
| Debilidad muscular | X |
| Cataratas | |
| Sordera de cualquier tipo | |
| Neuropatía sensitiva axonal | |
| Depresión | |
| Hipogonadismo | |
| Parkinsonismo | |
| Ataxia cerebelosa | |
| Pie cavo | |
| Temblor | |
| Discapacidad intelectual | |
| Rabdomiólisis por alcohol | |
| Oftalmoplejía | X |
| Antecedentes familiares de CPEO | X |
| Lactoacidosis | |
| Intolerancia al ejercicio | |
| Retinopatía pigmentaria |
Fuente: elaboración propia.
La CPEO se manifiesta con síntomas leves en la niñez, tal como ocurrió en el paciente que reportamos (16). La debilidad muscular ocular se desarrolla gradualmente durante años e incluso décadas. Puede ocurrir por mutación esporádica (deleción única), herencia mitocondrial (matrilínea) o ser autosómica dominante o recesiva 7.
Algunas mutaciones de POLG se han encontrado con efecto tanto dominante como recesivo, y la variabilidad clínica para homocigotos es muy variable. Se ha sugerido que las manifestaciones clínicas se ven influenciadas por factores modificadores como la composición del ADN mitocondrial, otros genes nucleares, factores epigenéticos y factores ambientales. Aquí se encontró en el paciente reportado la mutación p.Ala467Thr en estado de homocigosis, sugiriendo una herencia autosómica recesiva. Aunque los padres no tenían la secuenciación para el gen en cuestión, si la madre y el padre fueran portadores de la mutación, la mutación tendría un comportamiento recesivo. Otra posibilidad es que la madre sea portadora y el padre homocigoto, caso en el cual, la mutación también se heredaría en forma autosómica recesiva. Esta situación ilustra la complejidad en la herencia de esta patología, y justifica la necesidad de la evaluación molecular de los padres y la valoración por genetista para una consejería genética adecuada 10.
Los ensayos terapéuticos realizados hasta el momento no han sido exitosos. Dos grandes revisiones clínicas no encontraron evidencia que demuestre el beneficio de los agentes terapéuticos o suplementos nutricionales 1,17,18.
CONCLUSIÓN
La neumopatía crónica aspirativa es una complicación grave de las enfermedades neuromusculares, es imprescindible su reconocimiento temprano e inicio oportuno de intervenciones para retardar el deterioro de la función pulmonar en los pacientes con enfermedades neuromusculares. Ante el hallazgo de neumopatía crónica aspirativa se debe sospechar de la posibilidad de una enfermedad neuromuscular de base, es indispensable un equipo multidisciplinario para el diagnóstico, seguimiento y la asesoría genética.

