











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Kabuki es una patología de herencia autosómica dominante, debida a variantes en el gen KMT2D (OMIM # 147920) localizado en el cromosoma 12 (12q13.12) 1,2 con una prevalencia de 1 en cada 32.000 nacidos vivos 1-3. Se describió por primera vez en la población infantil nipona. Es un síndrome congénito de retardo mental con características asociadas como el enanismo posnatal, facie alargada con fisuras palpebrales largas y eversión del tercio lateral de los párpados inferiores. Los individuos tienen además una nariz ancha con punta nasal deprimida, lóbulos de las orejas prominentes, paladar hendido o en ojival, escoliosis, quinta falange de la mano corta, persistencia de almohadillas en los pulpejos de los dedos, recurrencia de otitis media en la infancia, anormalidades radiográficas en las vértebras, manos y cadera 1-3.
Se han publicado alrededor de 400 casos a nivel mundial, en Colombia se encuentran 6 publicados de este. El compromiso facial, las anomalías esqueléticas, las alteraciones en los dermatoglifos, la talla baja y el retraso mental fueron las principales características descritas de los pacientes con este síndrome en 1988.
Se han planteado otras manifestaciones como las fisuras palpebrales largas, eversión del párpado inferior, ptosis palpebral, cejas arqueadas y despobladas en el tercio externo, pestañas largas, estrabismo, escleras azules, puente nasal deprimido, labio superior fino e inferior grueso, dentición anómala, paladar fisurado o labio leporino, micrognatia, pabellones auriculares prominentes o malformados, hipoacusia, almohadillas en el pulpejo de los dedos, braquidactilia, escoliosis, hemivértebras, cardiopatías, malformaciones renales, criptorquidia, deficiencia en la hormona del crecimiento, hipotonía, entre otros 1-4. Existe una variante llamada síndrome de Kabuki tipo 2, con mutaciones en el gen KDM6A (OMIM # 300867) y de características clínicas parecidas al síndrome de Kabuki mencionado (tipo 1), pero cuyo patrón de herencia está ligado al cromosoma X (Xp11.3) 1,2.
Se reporta un caso pediátrico de síndrome de Kabuki. El presente trabajo busca fomentar el reconocimiento del fenotipo asociado al síndrome de Kabuki, con el fin de facilitar su diagnóstico oportuno a través del reporte de un caso. Toda la información referente al caso fue proporcionada por los familiares del paciente, quienes aceptan compartir su información asentida previamente y avalada por el Comité de ética de la Universidad del Norte en Barranquilla, quien avaló la realización de este reporte de caso de igual manera.
CASO CLÍNICO
Paciente masculino con 9 años de edad, natural y residente de Barranquilla (Colombia), producto de una segunda gestación de 33,1 semanas. Nacido por cesárea segmentaria transperitoneal indicada de acuerdo con una ruptura prematura de las membranas y la bradicardia fetal. Los familiares del paciente no refirieron antecedentes familiares de ningún tipo y la gestación de la madre no presentó alteraciones relevantes.
En el nacimiento pesó 2.300 gramos, su longitud y el perímetro cefálico fueron de 45 y 31 centímetros, respectivamente. Hallaron un implante bajo las orejas, paladar ojival, cuello alado, hipertelorismo, hipotonía y criptorquidia derecha, por lo cual es sometido a una intervención quirúrgica posteriormente. Posteriormente se identificaron nuevos hallazgos clínicos tales como: riñón derecho ectópico, síndrome bronco obstructivo, neumonías a repetición, laringotraqueítis, pansinusitis, otitis medias recurrentes, convulsiones febriles, anemia microcítica e hipocrómica, reflujo gastroesofágico, gastroduodenitis, displasia de cadera, hepatitis, somatomedina C disminuida, infección por molusco contagioso, piodermitis, infecciones de las vías urinarias en varias ocasiones quiste pancreático, dengue, varicela, piodermitis, historia de herpes positiva, infección por virus de Epstein-Barr. Tiene variante probablemente patogénica para síndrome de Kabuki tipo 1 por estudio genético (Tabla 1).
Tabla 1 Descripción de los estudios genéticos realizados al paciente
| Gen/Transcrito | Cambio nucleótido / Proteína | Exón/Efecto | Cigosidad/Frecuencia variante | Categorización de la variante |
|---|---|---|---|---|
| KMT2D NM_003482.3 | c.14918_14919insA p.(Asp4973Glufs*34) | Exón 48 Frameshif insertion | Heterocigota 49 % | Probablemente patogénica |
Resultados obtenidos de la historia clínica. Fuente: creación propia
Los hallazgos positivos del examen físico mostraron características craneofaciales definidas (Figura 1), tales como: cejas arqueadas con tercio lateral despoblado, pestañas alargadas, fisuras palpebrales elongadas, coloración ligeramente amarilla de la esclera bilateralmente, puente nasal deprimido, boca en carpa, paladar ojival, dentición anormal, pabellón auricular de baja implantación y orejas prominentes. Por otra parte se observó almohadillas en el pulpejo de los dedos (Figura 2), clinodactilia, braquidactilia (Figura 3), articulaciones laxas e hipermóviles y talla baja.
Se realizan estudios genéticos e inmunológicos para sustentar la sospecha diagnóstica clínica; también para buscar inmunodeficiencia debido a las múltiples infecciones presentadas por el paciente. A través de la obtención de una muestra de sangre periférica del paciente se procede a la extracción de ADN para la realización de ExoNIM clínico, es decir, una secuenciación del exoma, dirigido a la identificación de ciertas variantes en 5.713 genes que pueden estar asociados de forma definida al fenotipo del paciente. El estudio de secuenciación masiva se realizó utilizando un kit de captura de NimbleGen diseñado a medida y un secuenciador de la plataforma HiSeq de Illumina.

Figura 1 Rasgos craneofaciales. Figura obtenida previa autorización del paciente. Fuente: creación propia
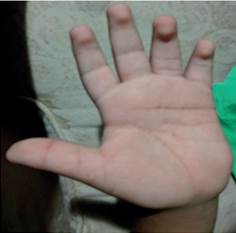
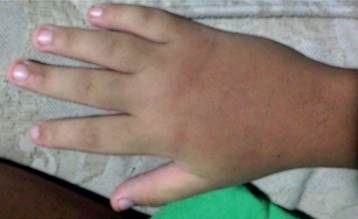
Los hallazgos genéticos e inmunológicos en detalle se presentan a continuación (Tabla 2 y 3) junto con su interpretación.
Tabla 2 Pruebas inmunológicas
| Prueba | Resultado | Valor de referencia |
|---|---|---|
| Linfocitos B: CD19+, CD20+ | 258,5 cel/ul | 600-2.700 cel/ul |
| Linfocitos CD4 | 562/mm3 | 544 a 1.663/mm3 |
| Linfocitos CD8 | 1.226/mm3 | 272 a 932/mm3 |
| Linfocitos T CD3 (totales) | 1.887/mm3 | 959 a 2577/mm3 |
| Relación CD4/CD8 | 0,46 | 0,93 a 4,50 |
| Linfocitos totales (CD 45+) CD3 % CD4 % CD8 % | 2347 80 % 24 % 52 % | |
| Inmunoglobulina A | 58,8 mg/dL | 21-291 mg/dL |
| Inmunoglobulina G | 1.248 mg/dL | 540-1.822 mg/dL |
| Inmunoglobulina M | 155,2 mg/dL | 41-183 mg/dL |
| Inmunoglobulina D | Menor de 4,06 mg/dL | 0 a 152,7 mg/dL |
| Complemento hemolítico total (CH-50) | Menor de 265,0 unidades | 392 a 1019 |
Resultados obtenidos de la historia clínica. Fuente: creación propia
Tabla 3 Electroforesis de proteínas
| Electroforesis de proteínas | ||
|---|---|---|
| Proteína | Resultado | Valor de referencia |
| Albúmina | 58,2 % | 55,8-66,1 % |
| Alfa 1 | 4,6 % | 2,9-4,9 % |
| Alfa 2 | 12,5 % | 7,1-11,8 % |
| Beta 1 | 5,9 % | 4,8-7,2 % |
| Beta 2 | 3,7 % | 3,2-6,5 % |
| Gamma | 15,1 % | 11,1-18,8 % |
| Albúmina C | 41,6 gr/L | 40,2-47,6 gr/L |
| Alfa 1_C | 3,3 gr/L | 2,1-3,5 gr/L |
| Alfa 2_C | 8,9 gr/L | 5,1-8,5 gr/L |
| Beta 1_C | 4,2 gr/L | 3,4-5,2 gr/L |
| Beta 2_C | 2,6 gr/L | 2,3-4,7 gr/L |
| Gamma C | 10,8 gr/L | 8,0-13,5 gr/L |
Se observa un aumento en la región Alfa 2 y posible componente monoclonal en la región Gamma. Resultados obtenidos de la historia clínica. Fuente: creación propia
DISCUSIÓN
Aunque es relativamente poco frecuente, el síndrome de Kabuki es una entidad cuyo diagnóstico es muy importante, por su impacto sobre la morbimortalidad y el desarrollo psicosocial de quien lo padece, además de las complicaciones derivadas de la enfermedad. Son múltiples y variadas las manifestaciones físicas y psicomotoras de la patología, por lo que reportar estos casos permite a la comunidad científica reconocerlas oportunamente y brindar asesoría a la familia del o de la paciente 2.
El síndrome de Kabuki se distingue por anomalías faciales que al identificarse oportunamente, llevan a la sospecha diagnóstica, que es principalmente clínica.
Cabe resaltar que las características consideradas como principales en el síndrome se encontraron presentes en el caso descrito, tales como el compromiso facial (cejas arqueadas con tercio lateral despoblado, fisuras palpebrales elongadas, puente nasal deprimido, pabellón auricular de baja implantación), las alteraciones en los dermatoglifos, la talla baja y el compromiso del neurodesarrollo, indicando un fenotipo distintivo que permite la identificación o, al menos, la sospecha de esta genopatía 1-4.
Es recomendable realizar, inicialmente en los pacientes con sospecha del síndrome de Kabuki estudios genéticos que revelen variantes genómicas relacionadas con este. La presencia de mutaciones en el gen KMT2D (localizado en el cromosoma 12q.13.12), se asocia con un patrón de herencia autosómico dominante del síndrome de Kabuki tipo 1. La variante identificada en la heterocigosis de la muestra del paciente descrito se trata de una inserción que provoca un cambio en la fase de lectura que no ha sido previamente registrada en la literatura ni en las bases de datos consultadas, está asociada con un fenotipo específico. A la espera de estudios funcionales o de la identificación de nuevos casos, la variante identificada se considera probablemente patogénica. Existe una variante llamada síndrome de Kabuki tipo 2, con mutaciones en el gen KDM6A y de características clínicas parecidas al síndrome de Kabuki mencionado (tipo 1), pero cuyo patrón de herencia está ligado al cromosoma X. El KDM6A está relacionado con la formación del corazón y las vértebras a partir del mesodermo, lo que pudiera generar un fenotipo distintivo.
La ausencia de cardiopatía en el paciente, si bien es llamativa, no significa un obstáculo para arribar al diagnóstico. Resulta interesante también la presencia de colangitis esclerosante primaria. Esta es una enfermedad sin etiología definida que causa fibrosis de las vías biliares intra y extrahepáticas. Se encuentra asociada comúnmente con la enfermedad de Crohn y a la colitis ulcerativa, ambas relacionadas con alteraciones del sistema inmune 5-8. La esclerosis de las vías biliares condiciona la obstrucción del flujo adecuado de bilis hacia el duodeno 5-8.
La obstrucción puede ser la causa de hipercolesterolemia, hipertrigliceridemia, hepatomegalia e hiperbilirrubinemia 9, relacionada con la formación de xantelasmas, xantomas 10 y el tinte ictérico. Debe recalcarse que los síntomas descritos, y otros encontrados, generan un impacto significativo sobre la calidad de vida del paciente 5-9,11. Esto es importante en su contexto, pues presenta, además, señales de alteración en el sistema inmune como infecciones a repetición.
Debe tenerse en cuenta, que las personas con síndrome de Kabuki presentan un mayor riesgo de contraer enfermedades inmunitarias, tales como el déficit de IgA, anemia hemolítica y púrpura trombocitopénica 1.
Sin duda la sospecha diagnóstica es meramente clínica y se alcanza con la simple observación y una detallada anamnesis. Consideramos imperativo fomentar el conocimiento de este síndrome, no solo entre los médicos especialistas que pueden involucrarse con los pacientes de acuerdo con cada manifestación clínica, sino entre los médicos generales y estudiantes, que constituyen el primer contacto con el sistema sanitario, a fin de posibilitar el diagnóstico oportuno y ofrecer a los cuidadores una guía sobre prevención, manejo, pronóstico y futuro de los pacientes.
CONCLUSIÓN
El síndrome de Kabuki es una patología de herencia autosómica dominante debida a variantes en el gen KMT2D localizado en el cromosoma 12 1,2. Se caracteriza principalmente por alteraciones faciales que inducen la sospecha diagnóstica. El paciente reportado presentaba múltiples de los hallazgos distintivos del síndrome descritos en la literatura. Si bien la variante identificada en heterocigosis en la muestra del paciente se trata de una inserción que provoca un cambio en la fase de lectura que no ha sido previamente registrada en la literatura ni en las bases de datos consultadas, está asociada con un fenotipo específico; está a la espera de estudios funcionales o de la identificación de nuevos casos, la variante identificada se considera probablemente patogénica para el síndrome de Kabuki tipo 1.

