











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La colangitis esclerosante primaria (CEP) es una enfermedad hepática colestásica crónica caracterizada por la inflamación y fibrosis de los conductos biliares, intra y extrahepáticos. La estenosis progresiva del árbol biliar lleva al desarrollo de secuelas como la cirrosis, hipertensión portal y enfermedad hepática terminal. Su etiología es desconocida, pero hay evidencia de una predisposición genética y mecanismos autoinflamatorios como componentes involucrados en la enfermedad (1,2).
En muchos casos los pacientes son asintomáticos, pero se pueden presentar ictericia, prurito, fatiga y dolor en el abdomen superior como los síntomas más comunes del fenotipo sintomático de la enfermedad (3). El diagnóstico se establece al visualizar dilataciones y estenosis multifocales en la vía biliar mediante colangiopancreatografía por resonancia magnética (CPRM). Hay una fuerte asociación con la enfermedad inflamatoria intestinal (EII), por lo que se requiere detección y vigilancia colonoscópica (4).
Es necesario distinguir la enfermedad de otro tipo de lesiones biliares como la colangitis bacteriana crónica, las lesiones isquémicas de las vías biliares por agentes como el formol o alcohol en el tratamiento de quistes hidatídicos, colangiopatía infecciosa asociada al síndrome de inmunodeficiencia adquirida, cirugía previa de las vías biliares y, lesiones congénitas y neoplásicas del tracto biliar (5).
Hasta la fecha no existe ningún tratamiento farmacológico aprobado. El trasplante hepático es el único manejo disponible con eficacia probada para evitar la progresión de la enfermedad (6). La farmacoterapia solo está enfocada en tratar los síntomas y manejar las complicaciones. Se han reportado tasas de supervivencia a los cinco años del 80 al 85 % postrasplante (7).
EPIDEMIOLOGÍA
La incidencia de la CEP está entre 0 y 1,3 casos por cada 100.000 personas/año. La prevalencia varía de 0 a 16,2 casos por cada 100,000 personas, siendo mayor en la población del hemisferio norte, como Norteamérica y Europa septentrional (8,9).
Si bien la CEP puede afectar a mujeres y hombres en cualquier época de la vida, presenta un predominio en los individuos masculinos con un 65-70 % de los casos y un pico de incidencia alrededor de los 40 años (10).
Se ha encontrado una importante asociación con la EII. Aproximadamente, el 70 % de los pacientes con CEP presentarán colitis ulcerosa en algún momento de la enfermedad, lo que sería un factor de riesgo independiente para desarrollar displasia colónica y cáncer colorrectal (1).
FISIOPATOLOGÍA
La CEP se ha clasificado como una colangiopatía idiopática, caracterizada por inflamación, fibrosis y colestasis (1). Actualmente, solo se cuenta con hipótesis sobre su fisiopatología y se ha sugerido que, después de una exposición ambiental aún no identificada, varias vías genéticamente predispuestas contribuyen a la lesión inmunológica persistente de los colangiocitos y al desarrollo de la enfermedad (10,11).
Los familiares de primer grado de los pacientes con CEP tienen un riesgo de 9 a 39 veces mayor de desarrollar esta enfermedad (12). Se han encontrado múltiples genes de susceptibilidad asociados, como los loci HLA-B8 y HLA-DR3, algunos de los cuales también están relacionados con la EII (10,13). Además, los genes de la vía de la interleucina 2 y las variaciones en el complejo mayor de histocompatibilidad clase I también se han asociado con la susceptibilidad a la CEP (11).
La mayoría de los factores ambientales que podrían contribuir a la CEP son desconocidos. Sin embargo, algunas teorías incluyen la dieta, el desequilibrio microbiano y las infecciones (10). No se ha encontrado una relación clara con el consumo de café, alcohol, cigarrillo o exposición a animales de granja (13).
El desequilibrio microbiano puede ocurrir a través de la dieta o de agentes infecciosos que pueden alterar la integridad intestinal, provocando así una inflamación y aumentando la exposición a bacterias y otros patógenos del sistema inmune. Esto se ha denomina-do la teoría del “Intestino permeable”, la cual ha sido apoyada por su relación con la EII (10,11). Incluso se ha demostrado que ambas enfermedades tienen perfiles intestinales microbianos similares (Veillonella, Enterococcus, Fusobacterium y Lactobacillus) (13).
Se piensa que los colangiocitos también pueden participar de forma activa en la patogénesis de la enfermedad al expresar citocinas proinflamatorias como el factor de necrosis tumoral alfa, interleucinas 6 y 8, entre otras, contribuyendo al reclutamiento de células inmunes y al desarrollo de la enfermedad (10,13).
Las principales células inmunes implicadas en la fisiopatología de la CEP son los linfocitos T, que inducen la senescencia en los colangiocitos y destrucción de las vías biliares (13). Este proceso puede acelerar la transformación neoplásica. La contribución de los linfocitos B no está clara, aunque se han descrito ni-veles séricos elevados de inmunoglobulina G4 (IgG4) en el 10 % de los casos (10).
La CEP también se ha relacionado con un componente autoinmune, en el que se ha encontrado el autoanticuerpo ANCA en más del 90 % de los pacientes, observado además en la colitis ulcerosa y en la hepatitis autoinmune (HAI). Sin embargo, este componente autoinmune se cuestiona por no cumplir con los caracteres epidemiológicos de la enfermedad (presentarse principalmente en mujeres), no responder a inmunomoduladores y no tener autoanticuerpos específicos (12).
La colestasis generada conduce a un ciclo de lesión progresiva en los colangiocitos debido a la composición tóxica de los ácidos biliares (11). Esto se ha demostrado al observar la mejoría histológica en los pacientes al bloquear la recaptación terminal de ácidos biliares (1,13).
Finalmente, la CEP debe considerarse como una enfermedad premaligna. Se ha visto que el 10-20 % de los pacientes con esta, desarrollan colangiocarcinoma, que puede ser la primera manifestación en los pacientes con CEP subclínica. También hay un mayor riesgo de cáncer extrahepático, incluido el de colon, vesícula biliar y páncreas. Curiosamente, el riesgo de contraer carcinoma hepatocelular parece ser menor en otras enfermedades hepáticas crónicas (1).
CLÍNICA
La estenosis y fibrosis progresiva de los conductos biliares producen colestasis considerable que termina por deteriorar el funcionamiento hepático. Alrededor del 15-55 % de los pacientes con CEP son asintomáticos y, en muchos de estos pacientes, el único indicio de la enfermedad es la alteración de los exámenes bioquímicos en donde se observa, predominantemente, un perfil colestásico; es decir, valores elevados de fosfatasa alcalina (FA) y gamma glutamil transferasa (GGT) (5). Debido a esto, el diagnóstico puede ser incidental o estar orientado por las alteraciones del perfil hepático y pocas veces por las manifestaciones clínicas (11).
Los síntomas son inespecíficos. Los más frecuentes son la fatiga y el prurito (5). También aparecen signos y síntomas como el dolor en el hipocondrio derecho, ictericia que incluso puede presentarse como hiperpigmentación, y otros menos comunes como la fiebre, escalofríos y pérdida de peso (5,10). En el examen físico los hallazgos más comunes son hepatomegalia y esplenomegalia (14). En las fases más avanzadas se hace evidente el deterioro del funcionamiento hepático. Como consecuencia de esto puede presentarse ascitis, sangrado por várices esofágicas y encefalopatía (1). La CEP se puede clasificar en varios subtipos definidos por la colangiografía y por histología. Estos subtipos son el clásico, el de conductos pequeños y la CEP asociada con la HAI. De acuerdo con la presentación de cada uno de estos se definen principalmente el pronóstico y algunas de las manifestaciones clínicas. La presentación clásica se da en el 90 % de los pacientes, es la de peor pronóstico, los otros subtipos se presentan en el 10 % restante, siendo ambos de mejor pronóstico que el subtipo clásico (10).
Una de las principales características de la CEP de presentación clásica es que suele acompañarse de EII, principalmente colitis ulcerosa. Esto se da aproximadamente en el 70-80 % de los pacientes, por lo cual, además de la disfunción hepática, también se presentarán síntomas gastrointestinales (15,16). Además, se han asociado en menor medida otras enfermedades como los cálculos y pólipos de la vesícula, osteoporosis, artropatías y otras enfermedades autoinmunes (10). Es importante el seguimiento a los pacientes, ya que esta enfermedad es considerada como un factor de riesgo para colangiocarcinoma (17).
DIAGNÓSTICO
Considerando que la CEP es una enfermedad, en la mayoría de los casos asintomática en sus primeras etapas, el diagnóstico suele hacerse en pacientes que presentan un patrón colestásico en el perfil hepático cuando la CPRM o la colangiopancreatografía retrógrada endoscópica (CPRE), muestran cambios característicos del conducto biliar (1). Los pacientes que tienen características histológicas compatibles con la CEP, pero con una colangiografía normal, se clasifican dentro del grupo de pacientes con la CEP de conducto pequeño (5 %) (18).
Aunque el principal hallazgo en el perfil hepático es un patrón colestásico, se puede observar también la elevación de las aminotransferasas, 2 y 3 veces por encima del valor normal, mientras que las bilirrubinas se encuentran dentro de los límites normales en la mayoría de los casos (6,11). Es importante tener precaución, ya que en estos casos podemos estar ante un síndrome de superposición de HAI/CEP. Por esto, un dato importante para sospechar de la CEP en pacientes asintomáticos con elevación de la FA es el antecedente de EII debido a que, como se mencionó previamente, suele aparecer como comorbilidad (6). Además, en un 10-20 % de los pacientes con la CEP se pueden observar niveles elevados de IgG4 (3,6). De ahí la recomendación de medir siempre los niveles de IgG4 al momento del diagnóstico de la CEP (7,19).
La medición de anticuerpos es particularmente importante cuando se sospecha del síndrome de superposición HAI/CEP. Sin embargo, para el diagnóstico de la CEP no son suficientes (11). Los pacientes con anticuerpos antinucleares, anticuerpos de músculo liso positivos y concentraciones elevadas del total de inmunoglobulinas deben alertar sobre la posibilidad de HAI, enfermedad relacionada con IgG4 o el síndrome de superposición HAI/CEP (3,6).
La CPRM es el método estándar para el diagnóstico de la CEP debido a su naturaleza no invasiva, ya que evita complicaciones potencialmente graves como la pancreatitis y colangitis bacteriana (20). En esta se observa un aspecto anular causado por estenosis multifocales cortas de los conductos biliares intrahepáticos (25 %), extrahepáticos (5 %) o ambos (70 %), con segmentos alternos normales o ligeramente dilatados, se ha descrito con aspecto en forma de grano de arroz. Posee una sensibilidad del 86 % y una especificidad del 94 % (18,21).
La CPRE se reserva únicamente para intención terapéutica o para evaluar estenosis en los conductos biliares, esto debido a su invasividad y al riesgo de presentarse complicaciones (21). Una ventaja importante de la CPRM es que permite obtener una mejor imagen de la zona proximal preestenótica, aunque lo hace en detrimento de una menor definición de los conductos biliares (6). Debido a esto, la Asociación Europea para el Estudio del Hígado (EASL) y la Asociación Americana para el Estudio de Enfermedades Hepáticas (AASLD) recomiendan la CPRM como el estudio imagenológico de primera elección para el diagnóstico de la CEP (18).
La ecografía de abdomen es útil para excluir litiasis biliar y para identificar hipertensión portal (20). Por su parte, la tomografía axial computarizada solo se utiliza para la estratificación y el diagnóstico del colangiocarcinoma, dada su baja sensibilidad (1).
La utilidad de la biopsia hepática en la CEP es controvertida. Se ha cuestionado mucho en el diagnóstico de la CEP de conducto grande, pero es valiosa cuando la CPRM es normal (22). Es útil para descartar las características superpuestas de la HAI y para diagnosticar la enfermedad de conductos pequeños (20). Las características histológicas clásicas se describen como una fibrosis periductal concéntrica, en forma de piel de cebolla. Sin embargo, es frecuente que estos hallazgos no se observen en las biopsias, especialmente, en estadios tempranos (1).
MANEJO FARMACOLÓGICO
No se ha comprobado una mejoría en la supervivencia en la CEP con el uso único del tratamiento médico (1). A pesar de que el beneficio del ácido ursodesoxicólico (AUDC) a largo plazo no está claro, se sigue utilizando en dosis moderadas de 15-20 mg/kg/día, con las cuales se ha visto una normalización en los niveles de FA, pero sin mostrar beneficios clínicamente relevantes en la mejoría de los síntomas, necesidad de trasplante o mortalidad (23-25). No se recomiendan dosis mayores debido a un mayor riesgo de cirrosis, várices y mortalidad (26). En pacientes sin respuesta terapéutica o intolerancia al AUDC puede considerarse el uso de ácido obeticólico, el cual ha demostrado mejoría en el perfil hepático a costa de mayores efectos adversos, especialmente, prurito. Otros fármacos se han sugerido para el manejo de la CEP, pero no se ha mostrado que mejoren el resultado en la enfermedad clásica (27). Entre estos se encuentran la terapia inmunosupresora, la combinación de AUDC con metronidazol o con fibratos y la vancomicina, la cual luce prometedora, al reducir los niveles de FA y bilirrubina, pero aún se requiere una mayor investigación (27,28-31). Cabe destacar la posible utilidad del trasplante de microbiota fecal para el manejo de la CEP, siendo seguro para estos pacientes, aunque se necesitan ensayos adicionales para determinar si tiene un papel por desempeñar en el tratamiento (32).
Para el manejo sintomático del prurito se cuenta con la colestiramina como primera línea (1). El prurito leve puede tratarse solo con emolientes y antihistamínicos (7). Como segunda línea, pueden usarse rifampicina o naltrexona (1). En el caso de la fatiga no existe una terapia recomendada debido a la dificultad de su tratamiento y evidencia limitada. No obstante, es importante excluir otras condiciones que puedan causar o empeorar el síntoma, como la depresión, anemia, insuficiencia suprarrenal, los trastornos del sueño o hipotiroidismo (3).
Los pacientes con la CEP tienen una incidencia de osteoporosis del 4-10 %, además, pueden tener deficiencia de vitamina D (33). Por esto, se recomienda realizar densitometría ósea al momento del diagnóstico y repetirla cada 2-4 años (7). En aquellos pacientes con la CEP y osteopenia confirmada se debe administrar calcio 1-1,5 gr/día y vitamina D 1000 UI/día, en caso de osteoporosis, se deben añadir bifosfonatos (10).
La colangitis aguda es una complicación común en la CEP, su manejo requiere descompresión del segmento biliar obstruido y el uso de antibióticos, preferiblemente, guiados por antibiograma. En caso de realzar el manejo empírico se deben usar agentes que cubran los principales patógenos biliares (E. coli, Klebsiella, Enterococcus y especies anaerobias), como ampicilina y sulbactam, ceftriaxona, metronidazol o fluoroquinolonas (34,35).
MANEJO ENDOSCÓPICO
El principal objetivo de esta terapia es descomprimir los segmentos biliares obstruidos por la enfermedad estenosante. La AASLD recomienda este tipo de manejo para los pacientes con síntomas de estenosis dominante: colangitis, ictericia, prurito o dolor en el cuadrante superior derecho del abdomen (36).
La principal indicación para la intervención endoscópica es la presencia de estenosis dominante, es decir, que haya un diámetro menor de 1,5 mm en el conducto biliar común o, menor de 1 mm, en los conductos hepáticos (37). Encontrar esto en un paciente implica un peor pronóstico con índices de supervivencia marcadamente reducidos (38).
Las guías primarias para la CEP recomiendan la dilatación con balón como tratamiento endoscópico de primera línea (Figura 1). Si se decide realizar cualquier intervención biliar endoscópica se debe estar alerta ante el riesgo de colangitis bacteriana y administrar siempre antibióticos profilácticos de acuerdo con los perfiles de resistencia locales (39). Siempre se debe evaluar la presencia de várices, ya que en los pacientes con la CEP el desarrollo de estas puede darse antes de que sea evidente la cirrosis (7).
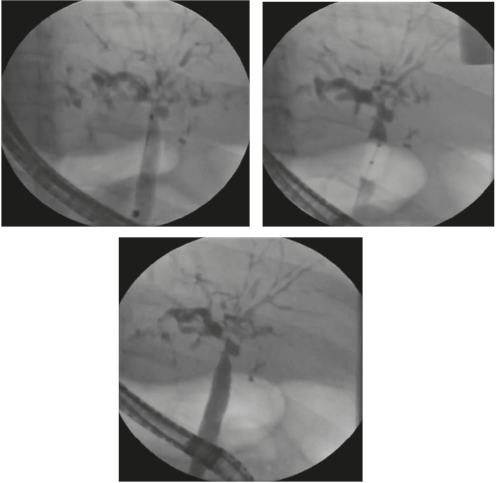
Figura 1 Secuencia de colangiografía con estenosis en la confluencia, angiograma con balón y cepillado.
Debido a la naturaleza invasiva del procedimiento, entre el 7,3 y el 20 % tienen riesgo de tener complicaciones posendoscópicas, como la pancreatitis, colangitis, perforación de la vía biliar y hemorragia (40).
TRASPLANTE HEPÁTICO (TH)
Sigue siendo el único tratamiento curativo para la CEP, aunque con el riesgo sustancial de una recurrencia de la enfermedad. El momento ideal para el TH no se conoce, pero está más influenciado por la disponibilidad de donantes de órganos que por la misma evidencia (1).
Las indicaciones para el TH incluyen a los pacientes con enfermedad hepática descompensada, prurito intratable, hipertensión portal refractaria a la terapia médica o colangitis bacteriana recurrente (41). Cabe anotar que el prurito intratable es indicación solo cuando se hayan agotado todas las opciones terapéuticas y excluido la comorbilidad psiquiátrica (3).
La tasa de supervivencia a 1 año, después del TH, su-pera el 90 % y, a los 5, es del 85 % (42). Al igual que ocurre con otras patologías que requieran del TH, el procedimiento tiene riesgo de desarrollar complicaciones como el rechazo celular agudo temprano (primeros 30 días), trombosis de la arteria hepática, estenosis biliar anastomótica y recurrencia. El rechazo agudo temprano responde bien a los corticoesteroides sistémicos y ha demostrado que no tiene ningún efecto sobre la supervivencia del injerto (3).
Al menos el 25 % de los pacientes trasplantados desarrollarán enfermedad recurrente 10 años después del trasplante (12). Su diagnóstico es de exclusión, después de descartar otras causas de estenosis en el aloinjerto, como el rechazo crónico, daño por reperfusión, isquemia biliar debido a la trombosis de la arteria hepática, incompatibilidad ABO, sepsis biliar o estenosis no anastomóticas tempranas (Tabla 1) (4). Entre los factores de riesgo para desarrollar recurrencia se encuentran la colitis ulcerosa, los factores genéticos, la gravedadde la CEP, infección por citomegalovirus y el rechazo celular agudo (4,43). No existe evidencia que respalde regímenes específicos de inmunosupresión postrasplante que tenga un efecto importante sobre la recurrencia (4). El aumento de las FA séricas puede indicar recurrencia, con la confirmación por CPRM, donde se observan las características típicas de la CEP (3). Un estudio encontró que la colectomía, antes y durante el TH inicial para la CEP, puede ser protectora contra la recurrencia (44).
Tabla 1 Criterios diagnósticos para la CEP recurrente después de un trasplante hepático
| Criterios de Inclusión | Criterios de exclusión |
| Diagnóstico confirmado de CEP* antes del trasplante hepático. Colangiografía: Estenosis biliares, no anastomóticas, intrahepáticas con arrosariamiento e irregularidades después de 90 días. Histología: Colangitis fibrosa o lesiones fibroobliterantes con o sin ductopenia, fibrosis biliar o cirrosis biliar. | Trombosis o estenosis de la arteria hepática. Rechazo crónico con ductopenia. Estenosis anostomóticas. Estenosis no anostomóticas antes de 90 días del postrasplate. Incompatibilidad ABO. |
*CEP: colangitis esclerosante primaria.
Fuente: adaptado de Graziadei, et al. (45)
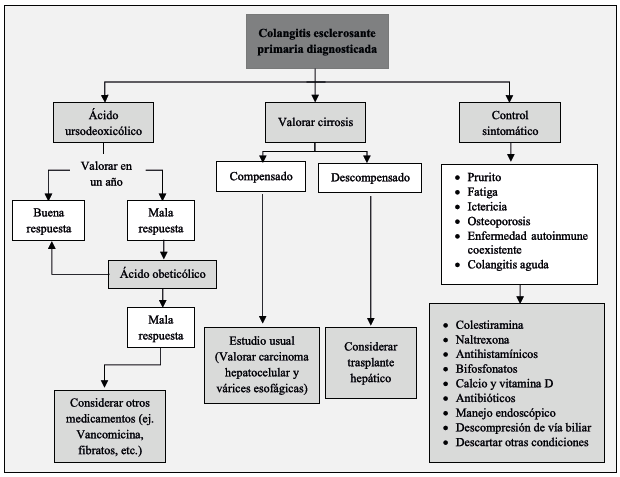
En la Figura 2 se esquematiza el manejo de la CEP.
SEGUIMIENTO Y PRONÓSTICO
El pronóstico general de los pacientes con la CEP sigue siendo pobre, con una supervivencia, sin TH, de 21 años. Se estima que en el 75 % de los pacientes la enfermedad progresará en los primeros 6 años posteriores al diagnóstico (11).
Las neoplasias hepatobiliares son una causa frecuente de mortalidad en estos pacientes. El riesgo de desarrollar colangiocarcinoma a lo largo de la vida es hasta del 20 %, con una incidencia anual de 1,5 % y una supervivencia a 5 años menor al 5 % (46). Los pacientes con estenosis dominante tienen mayor riesgo de desarrollar colangiocarcinoma; Por esto, la detección del cáncer es una parte clave del seguimiento de los pacientes con CEP (47).
La prueba estándar para las estenosis sospechosas es la citología por cepillado biliar junto con la hibridación in situ fluorescente (FISH), teniendo una sensibilidad del 68 % y especificidad del 70 %, aunque los costos limitan mucho su uso (45). Aun así, se recomienda utilizarla en pacientes con la CEP que tengan estenosis dominante vista por imagen para excluir el diagnóstico de colangiocarcinoma (7,47).
Las mediciones de antígeno de carbohidratos (CA19-9) y carcinoembrionario (CEA) pueden ayudar en el diagnóstico del colangiocarcinoma. Sin embargo, su baja sensibilidad y especificidad no permiten una detección efectiva (3,48). La CPRM combinada con la resonancia magnética contrastada es el estudio imagenológico mayormente utilizado para la evaluación anual, especialmente en pacientes con deterioro de la condición clínica o alteración del perfil hepático (Figura 3) (47).
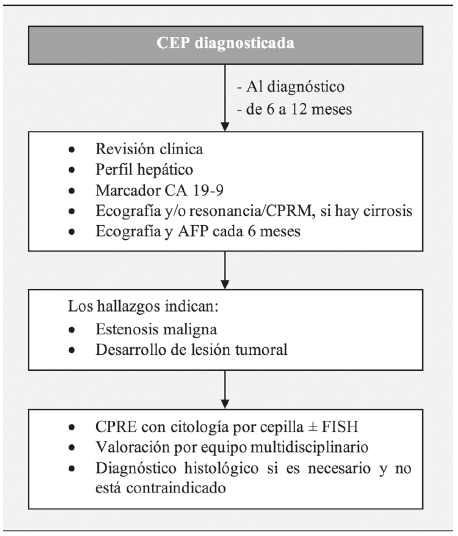
Fuente: adaptado de Karlsen et al. (49)
Figura 3 Algoritmo para la vigilancia del colangiocarcinoma en colangitis esclerosante primaria CEP: colangitis esclerosante primaria. CPRM: colangiopancreatografía por resonancia magnética. AFP: alfafetoproteína. CPRE: colangiopancreatografía retrógrada endoscópica. FISH: hibridación in situ fluorescente.
El riesgo de desarrollar cáncer de vesícula biliar es 10 veces mayor en pacientes con la CEP que en la población general. Los pacientes en quienes se encuentren pólipos en la vesícula biliar deben ser evaluados con ecografía cada 6 o 12 meses y, en general, se recomienda la colecistectomía si los pólipos son mayores de 8 mm. Sin embargo, la AASLD recomienda la colecistectomía independientemente del tamaño de la lesión (1).
La EII asociada con la CEP se relaciona con un mayor riesgo de contraer cáncer colorrectal (prevalencia aproximada de 60-80 %) (1). Una posible explicación para esta relación es que algunos de los medicamentos utilizados en la EII conducen a una absorción deficiente de folatos, esto se ha asociado con la alteración del proceso normal de metilación del ADN y cambios en el cromosoma y la cromatina (3). Las guías recomiendan realizar colonoscopia con biopsias de la mucosa anualmente en pacientes con EII y cada 5 años si no hay antecedente de EII (10). Es importante tener presente que el riesgo de aparecer cáncer colorrectal no se reduce después del trasplante, por lo que su vigilancia debe continuar posteriormente (3).
Sin TH la hepatopatía progresiva es una causa común de muerte. La cirrosis está presente en aproximadamente dos tercios de los pacientes que se someten a una biopsia de hígado. Se estima que, en el momento de la derivación inicial para la evaluación del TH, aproximadamente, del 30 al 50 % de los pacientes tendrán, mínimamente, fibrosis avanzada, un 25 % con várices esofágicas (11).
Los factores de buen pronóstico en la CEP incluyen el diagnóstico en edades tempranas, sexo femenino, niveles normales o bajos de FA, enfermedad de conducto pequeño y CEP con características de HAI. Entre los factores de pobre pronóstico están las extensas estenosis intra o extrahepáticas, estenosis dominantes, colangitis recurrentes, colitis ulcerosa y cirrosis con hipertensión portal (1). Es bien conocido que el empeoramiento de la colestasis predice peores resultados, independientemente de la presencia del trata-miento. Por esto, valores elevados de FA se asocian con un mayor riesgo de TH, cáncer biliar y muerte (11).
El modelo para la puntuación de la enfermedad hepática en la etapa terminal (MELD) es ampliamente utilizado para el pronóstico en los pacientes con cirrosis de cualquier etiología. Entre las personas con CEP, una puntuación MELD superior a 14 es un predictor fuerte de complicaciones clínicas relevantes, como la hemorragia por várices esofágicas, colangiocarcinoma, insuficiencia hepática, TH o muerte. Aun así, no parece predecir estos resultados mejor que la FA sola (11).
Se han desarrollado modelos predictivos específicos para la CEP, cada uno con fortalezas intrínsecas importantes. Sin embargo, la AASLD no recomienda el uso de modelos de pronóstico para la predicción de resultados en pacientes individuales, esto debido a la falta de consenso con respecto a cuál modelo funciona mejor (11).

