











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
La hipofosfatemia ligada al X (HL-X) se produce por mutaciones que generan inactivación del gen PHEX (en inglés: phosphate regulating endopeptidase analog, X-linked), lo que produce una pérdida renal de fosfato (1). A pesar de ser una entidad muy rara, es la más frecuente de los raquitismos hereditarios, con una incidencia de 1:20.000 nacimientos (2-4). Los niños afectados presentan raquitismo, deformidades óseas, reducción en la velocidad de crecimiento, talla baja final y defectos en la dentina. Por el otro lado, los adultos cursan con osteomalacia, abscesos dentales y formas de mineralización ectópica (5). Aunque la gravedad clínica es variable, es imperativo un diagnóstico oportuno con el fin de optimizar al máximo la talla final y minimizar las deformidades esqueléticas que conllevan a la presencia de complicaciones ortopédicas y mala calidad de vida en los pacientes afectados.
A continuación, se presenta un caso clínico del Hospital Napoleón Franco Pareja de la ciudad de Cartagena de Indias (Colombia) procedente del área rural, lo que dificultó el control del paciente para las visitas médicas; que fue remitido solo a los 14 años para ser evaluado por endocrinología y nefrología pediátrica. A esta edad se logra hacer un diagnóstico clínico y molecular que confirma el diagnóstico de HLX y se espera que el tratamiento mejore su calidad de vida, a pesar de haber sido realizado de manera tardía.
CASO CLÍNICO
Paciente masculino con 14 años procedente de una vereda del Carmen de Bolívar, fue remitido por una deformidad de los miembros inferiores del tipo genu varo a endocrinología pediátrica.
La madre no recuerda el peso ni talla al nacer. Niega antecedentes patológicos y quirúrgicos. Lactancia materna hasta los dos años. Como antecedente familiar, la madre presenta talla baja, alteraciones dentarias y deformidades en los miembros inferiores. El paciente tiene 4 hermanos (3 hombres y 1 mujer) sanos. Las deformidades del paciente en los miembros inferiores se hacen evidentes desde los 13 meses de vida, pero de acuerdo con el distanciamiento de su vereda de los centros de salud, así como las dificultades económicas y sociales, no se habían realizado los controles adecuados.
Los resultados del examen físico del paciente en el ingreso (edad 14 años con 10 meses) fueron: talla 137 cm (DE -3,92), peso 46 kg (DE + 1,54), relación SS/SI (segmento superior/segmento inferior): 1,14. Piezas dentales en mal estado, miembros inferiores en varo y distancia intercondílea de 20 cm. En la Figura 1 se observa al paciente con su deformidad ósea importante. Por este motivo, endocrinología pediátrica solicita estudios bioquímicos y hormonales complementarios por la sospecha de raquitismo, los cuales se encuentran en la Tabla 1. Se muestra como hallazgo significativo la hipofosfatemia asociada con la disminución de la reabsorción tubular de fosfatos (RTP).
Tabla 1 Laboratorios iniciales del paciente pediátrico
| Laboratorio | Resultados | Valores de referencia |
|---|---|---|
| Fósforo sérico | 1,93 | 3-5,4 mg/dl |
| Calcio sérico | 9,1 | 8,5-10,5 mg/dl |
| Fosfatasa alcalina | 353 | < 390 U/L |
| PTH | 38,7 | 10-60 pg/ml |
| 25 OH Vitamina D | 34,9 | > 30 ng/ml |
| 1,25 (OH)2 Vitamina D | 94,7 | 19-83 pg/ml |
| Fosfatasa alcalina | 353 | 30-390 U/L |
| RTP | 81,63 % | > 85 % |
| Tmp/GFR | 1,7 | 2,9-6,5 mg/dl |
| Gases arteriales | Normal |
PTH: hormona paratiroidea. RTP: reabsorción tubular de fósforo. TmP/GFR: reabsorción tubular máxima de fosfato por rata de filtración glomerular. Fuente: creación propia.
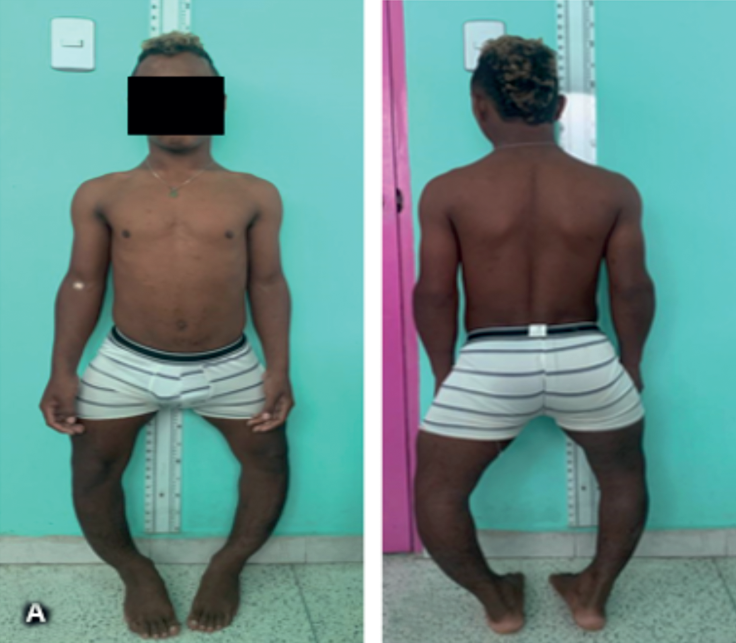
Fuente: tomado de la historia clínica del paciente con autorización
Figura 1 Fotos del paciente pediátrico reportado A. Imagen anterior. B. Imagen posterior donde se puede observar la deformidad importante en los miembros inferiores: genu varo.
Por su genu varo se solicita además radiografías de cadera y huesos largos (Figura 2), con hallazgos compatibles de raquitismo. Se solicita ecografía renal y vías urinarias con las que se descarta nefrocalcinosis. Sin olvidar el diagnóstico clínico y radiográfico del raquitismo hipofosfatémico, endocrinología pediátrica inicia manejo con sales de fosfato y calcitriol.
Se interconsulta al servicio de nefrología pediátrica para apoyo. Este solicita el panel de estudio molecular para raquitismo hipofosfatémico, halla una mutación del gen PHEX: variante c.733-2A>T. Por las manifestaciones clínicas severas, se considera iniciar anticuerpo monoclonal contra FGF-23 (burosumab).
La mamá, acompañante frecuente en la consulta de su hijo, presenta talla baja grave (talla 140 cm), deformidad en varo en miembros inferiores, abscesos dentarios múltiples, caídas de piezas dentarias (no tiene caninos ni incisivos superiores e inferiores). Por la clínica de la madre del joven, también se le realiza un estudio molecular: genotipificación del gen PHEX, se comprueba que presenta la misma mutación de su hijo (c.733-2A>T).
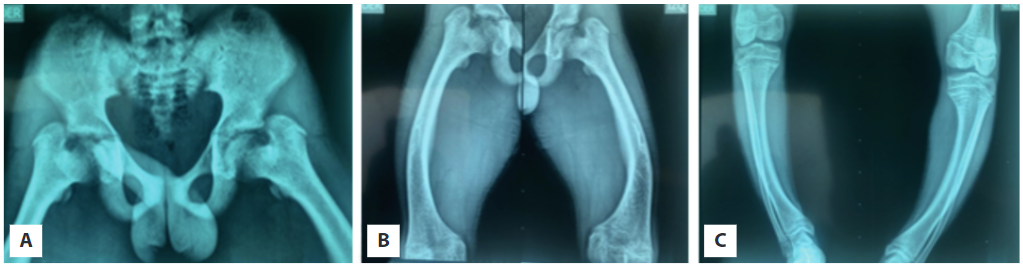
Fuente: tomado de la hisotira clínica del paciente con autorización
Figura 2 Radiografías de la cadera y los miembros inferiores A. Se observa el acortamiento del cuello femoral en forma bilateral, con ángulo cervicodiafisario de 150°, bilateral para coxas valgas. B. Metáfisis ensanchadas, fémur incurvado con pseudofractura en la cara lateral de ambos fémures. C. Miembros inferiores con deformidad en varo.
CONSIDERACIONES ÉTICAS
Se siguieron las normas sobre aspectos éticos de la investigación en seres humanos. Fue aprobado por el Comité de ética médica del Hospital y se contó con el consentimiento informado. Se conservó la confidencialidad de los pacientes de este reporte.
DISCUSIÓN
El raquitismo hipofosfatémico ligado al X (XLH) es un trastorno hereditario dominante, causado por mutaciones inactivantes del gen PHEX (Xp22.1), que codifica para una metaloproteasa, miembro de la familia M13 dependiente de Zinc, expresada en osteoblastos, odontoblastos y, en menor grado, en el pulmón, ovario, testículos y glándula paratiroides, que tiene como función modular la degradación del factor de crecimiento del fibroblasto-23 (FGF-23) (6-8). Se han reportado aproximadamente 460 tipos de mutaciones (9) que en el 20 % de los casos pueden ser espontáneas (7,10), como en el presente, donde la madre del paciente pediátrico, que no tiene antecedentes en los miembros de su familia, presenta una mutación espontánea o de novo que es trasmitida a uno de sus hijos. Se hace esta publicación debido a que es una entidad muy rara en el mundo y en este caso es más inusual, por ser una mutación espontánea. Además, llama la atención la edad tardía en que se hace el diagnóstico, tanto de la madre como del hijo, a pesar de tener manifestaciones clínicas muy sugestivas de raquitismo.
El FGF-23 induce la pérdida renal del fosfato al suprimir la expresión de cotransportadores sodio-fosfato (NaPi-2a y NaPi-2c) en la superficie apical del túbulo proximal de la nefrona, condicionando el desarrollo de hiperfosfaturia (11). FGF-23 también inhibe la acción de la 1α-hidroxilasa, necesaria para el paso de 25-Hidroxivitamina D a su forma activa 1,25 dihidroxivitamina D3 (1,25-OH-2D3), e igualmente reduce la absorción del fosfato del intestino y el hueso (12). Una pérdida sostenida de fosfato lleva a una profunda hipofosfatemia, alteración bioquímica fundamental que puede estar presente desde el nacimiento o después de los 6 meses de vida (13). Cuando aparece, se retarda el crecimiento esquelético y, en ocasiones, la edad ósea (14). Manifestaciones clínicas como talla baja y las lesiones óseas raquíticas son hallazgos fenotípicos característicos que están presentes en los pacientes de nuestro reporte, aunque su gravedad es muy variable entre los individuos que presentan esta entidad. En este reporte se observa claramente cómo el impacto social y económico puede influir en la vida de estos pacientes, al producir secuelas físicas y un compromiso psicológico muy evidente en el niño que ocasionan la desescolarización. Las radiografías de los huesos largos del paciente pediátrico exhiben una mineralización disminuida en medio de trabéculas escleróticas gruesas, ensanchamiento de metáfisis y arqueamiento de las piernas (14). Las enfermedades dentales como los abscesos radiculares, antecedentes presentes en la madre del joven, a menudo se desarrollan debido a los defectos en la dentina y el esmalte (15). Sin terapia, el crecimiento lineal se desacelera entre los 4 o 5 años de edad, presentándose talla baja evidente en la madre y el hijo (5,14). La entesopatía mineralizante y la osteoartropatía con frecuencia se desarrollan en la edad adulta (5).
En el seguimiento por nefrología pediátrica se solicitó el panel genético para raquitismo. Se encontró la mutación (c.733-2A>T) en el gen PHEX y se procedió a realizar solo la genotipificación del gen PHEX a la madre, lo que comprobó que tenía la misma mutación que su hijo. Se han utilizado diversos métodos moleculares para detectar mutaciones del gen PHEX, como la secuenciación directa de productos de PCR (reacción en cadena de la polimerasa), amplificación de sonda dependiente de ligadura múltiple (MLPA) y el análisis de ARNm de PHEX (11). Aunque estas pruebas no siempre se requieren para el diagnóstico de XLH; en casos donde no se tiene historia familiar de la enfermedad pueden ser necesarias como lo fue en la situación de nuestros pacientes.
En el caso de pediatría es evidente la mineralización ósea inadecuada y los defectos en la dentina, esto conlleva a las características clínicas distintivas como la talla baja, las deformidades esqueléticas de los miembros inferiores y las piezas dentales en mal estado.
A nivel radiológico se encuentra una pérdida de la definición y nitidez de la línea metafisaria distal con desflecamiento o “deshilachamiento”, pseudofracturas en la cara lateral de ambos fémures; asociados a la hipofosfatemia se observa disminución en la RTP y TmP/GRF. A pesar de existir una importante gravedad clínica, cabe anotar que los niveles séricos del fosfato en el niño no descendieron proporcionalmente, incluso con previas elevaciones en fosfatasa alcalina, lo que corresponde con los diversos fenotipos de la enfermedad. Además, es importante resaltar la ausencia de dichas características en otros familiares, por lo que se considera que se trata de una probable mutación espontánea en la madre y que ha sido trasmitida a su hijo.
Nuestro paciente pediátrico recibió, de forma irregular, el manejo tradicional que incluye vitamina D activada y soluciones de fosfato oral, que si bien es eficaz para aumentar el crecimiento infantil, es poco tolerada y los largos periodos del tratamiento aumentan el riesgo de hiperparatiroidismo secundario, nefrocalcinosis, nefrolitiasis y compromiso de la función renal (11,16), por lo que se optó por una terapia más novedosa y de mejores resultados. En el 2018, la EMA (European Medicines Agency) y la FDA (Food and Drug dministration) aprobaron el uso de burosumab en niños, un anticuerpo monoclonal tipo IgG1 humana recombinante, diseñado para reducir la actividad de FGF-23 (5).
Se administra por vía subcutánea y puede incrementar la TmP/GFR, reabsorción tubular de fósforo, los niveles de fosfato y 1,25-OH-2D3 séricos (5,9). Hasta el momento, los ensayos clínicos han demostrado el perfil de seguridad y eficacia del burosumab al mejorar el crecimiento lineal, la función física y reduciendo el dolor y la severidad del raquitismo, por lo que se incluye en el algoritmo de manejo de pacientes con XLH (17-20). Aún en nuestro país, el anticuerpo monoclonal contra FGF-23 no cuenta con registro sanitario en el sistema de salud, pero puede ingresarse al país bajo la modalidad de medicamento vital no disponible, regulado en el Decreto 481 de 2004 del INVIMA (Instituto Nacional de Vigilancia de Medicamentos y Alimentos). Se obtuvo la aprobación para utilizar esta terapia más novedosa y efectiva que el tratamiento tradicional.
Con este reporte de caso se pueden resaltar 3 aspectos que son de suma importancia. Primero, en verdad el HLX es una enfermedad rara y es posible que un médico nunca tenga la oportunidad de diagnosticarla, pero es importante que siempre que se presenten manifestaciones clínicas características, además de la hipofosfatemia, se piense en esta entidad. Segundo, los pacientes con HLX necesitan de un equipo interdisciplinario (no solo en el campo médico, sino también social y psicológico) que logre vencer las dificultades administrativas, económicas y sociales, con el fin de realizar un seguimiento integral desde el momento en que se sospecha de esta enfermedad, para evitar que aparezca la gran variedad de manifestaciones clínicas como en los casos reportados; algunas generan incapacidad y desescolarización. Por último, en la actualidad se cuenta con una estrategia novedosa en el tratamiento del HLX, esta ha mostrado resultados clínicos satisfactorios reflejados en los estudios del burosumab en niños y adultos.
CONCLUSIÓN
El raquitismo hipofosfatémico continúa imponiendo un importante reto diagnóstico y terapéutico debido a las consecuencias sobre el crecimiento y desarrollo esquelético, con el consecuente impacto sobre la función física. Por lo anterior, se requiere de un adecuado abordaje diagnóstico, con detección oportuna de los casos, para poder iniciar el manejo temprano de las alteraciones metabólicas, evitar el compromiso importante de la funcionalidad y mejorar la calidad de vida de los pacientes que padecen esta enfermedad.

