











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El complejo de Carney (CC) es un síndrome raro, de herencia autosómica dominante. Clásicamente descrito en 1985 por J. Aidan Carney como la combinación de lentiginosis, endocrinopatía y mixomas atriales (1). Esta entidad involucra múltiples órganos y las manifestaciones pueden ser variables. Se han implicado varios loci en el complejo: uno en el cromosoma 17q22-24 (CNC1) que codifica la subunidad reguladora de la proteína quinasa A (PRKAR1A), responsable del 70 % de los casos, y otro en el cromosoma 2p16 (CNC2) con un fenotipo muy similar (2). Recientemente se han descrito mutaciones en los genes PRKACA en el cromosoma 19p13.12 y PRKACB en el cromosoma 1p31.1 (3). La acromegalia es una de las endocrinopatías q ue se presentan con una menor frecuencia en estos pacientes, así como el schwannoma melanocítico. En el presente trabajo se describe un caso con estas patologías dentro del contexto de un CC y se hará una revisión de los aspectos clínicos más importantes de este síndrome. La paciente dio su consentimiento informado y se cumplieron los protocolos éticos de acuerdo con la Declaración de Helsinki (4).
Descripción del caso
Mujer de 31 años, desde la adolescencia con acné e hirsutismo; diaforesis, crecimiento acral, con cambio de talla del zapato de 38 a 40, cambio del tono de la voz, hipertensión y disnea de esfuerzos. Con sus cambios físicos se sospechó de una acromegalia. Se le hicieron estudios de laboratorio que confirmaron el diagnóstico (Tabla 1) y una resonancia de hipófisis en la que se documentó un macroadenoma en la silla turca (Figura 1). Se realizó resección transesfenoidal del adenoma pituitario en julio del 2012, requiriendo de una segunda intervención por persistencia de la actividad de la enfermedad y un tumor residual en noviembre del 2012. En la evaluación clínica se evidenciaron léntigos en la mucosa oral y mixomas cutáneos, presentes desde la infancia (Figura 2). Con estos hallazgos se planteó como posibilidad diagnóstica un complejo de Carney. Se solicitaron estudios adicionales buscando otras manifestaciones características de la entidad. Se hizo biopsia de una lesión en la mucosa labial, con resultado de patología que reportó lentiginosis, y de un nódulo en el brazo compatible con un mixoma. En la ecocardiografía no se encontraron mixomas cardiacos. En la ecografía de cuello se documentó un bocio multinodular con predominio de lesiones quísticas, ninguna de ellas sólidas o sospechosas de malignidad.
Tabla 1 Perfil hormonal de la paciente
| Hormona Evaluada Unidades | Inicial | Post Primera CX TE Jul/12 | Post Segunda CX TE Dic/12 | Noviembre 2013 | Diciembre 2017 | Marzo 2020 |
|---|---|---|---|---|---|---|
| Somatomedina C ng/mL | 720 (80-277) | 62 (80-277) | 504 (80-277) | 284,3 (80-245,7) | 276 (80-277) | 170 (63-223) |
| Somatropina (GH) ng/mL | Basal: 34,4; 30 min: 95; 60 min: 97,2; 90 min: 96,8 | 18,2 | 4,47 | 3,1 | 1,2† | 2,4† |
| TSH uUI/ml | 1,2 | 0,9 | 1,6 | NR | 1,3 | NR |
| Tiroxina libre ng/dL | 0,8 | 0,8 | 0,9 | NR | 1,1 | NR |
| Prolactina ng/mL | 17,6 | 11,8 | 25,8 | NR | 17,5 | NR |
| Cortisol ug/dL | NR | 4,7 | 8,7 | 8am: 10,4; 4pm: 3,6 | 8am: 9,3; 4pm: 4,2 | 7,6 |
| FSH mUI/mL | 3,5 | NR | NR | NR | 5,1 | NR |
| LH mUI/mL | 0,7 | NR | NR | NR | 5,9 | NR |
| ACTH pg/mL | NR | NR | NR | < 5 | 10,4 (0-46) | NR |
| Cortisol post DXMDB ug/dL | NR | NR | NR | 3,1* | NR | NR |
| Dehidroepiandrosterona ng/mL | NR | NR | NR | NR | NR | 1,9 (1,3-9,8) |
ACTH: Hormona adrenocorticotrópica, CX TE: cirugía transesfenoidal, DXMDB: Dexametasona a dosis bajas (1mg). FSH: Hormona Folículo estimulante, LH: Hormona Luteinizante, NR: No realizado, TSH: Hormona estimulante del tiroides. *Un valor mayor de 1.8 ug/dl asociado a una ACTH menor de 5 pg/ml confirma el diagnóstico de síndrome de Cushing de origen adrenal. † En acromegalia se recomiendan valores menores de 1ng/ml. Valores menores de 2.5 ng/ml se asocian a menor mortalidad respecto a valores mayores a 2.5 ng/ml. Fuente: creación propia.

Figura 1 Hallazgos imaginológicos. a. Macroadenoma hipofisiario de 22 x 27 x 20 mm; b. Imagen del adenoma adrenal izquierdo en la resonancia magnética de abdomen; c. Lesión mediastinal posterior, con elevación del hemidiafragma y calcificaciones en su interior y compromiso del bronquio fuente izquierdo.
En el seguimiento inicia con dolor pleurítico en hemitórax izquierdo e hipoxemia; se le realiza una radiografía de tórax donde se observa una opacidad basal izquierda y se caracteriza con tomografía de tórax contrastada, donde se encuentra una lesión mediastinal posterior (Figura 1). Cirugía de tórax la lleva a toracotomía encontrando un schwannoma melanocítico psamomatoso.
En el estudio tomográfico de tórax también se identificaron varios nódulos mamarios, con biopsia de uno de ellos compatible con un fibroadenoma, sin variación en el seguimiento ecográfico posterior.
Se realizó una tomografía de abdomen, la cual demostró un adenoma suprarrenal izquierdo (Figura 1). Se le efectuaron pruebas de laboratorio compatibles con un síndrome de Cushing de origen adrenal (Tabla 1), a pesar de que no presentaba ningún hallazgo fenotípico de esta entidad. Se practicó una adrenalectomía unilateral. La patología informó una hiperplasia nodular pigmentada. Recibió suplencia con glucocorticoides con desmonte posterior, sin evidencia bioquímica o clínica posterior de hipercortisolismo.
Para el manejo de la acromegalia la paciente requirió de tratamiento farmacológico con dosis altas de análogo de somatostatina (180 mg mensuales de Lanreotide) en combinación con un agonista dopaminérgico (cabergolina); lo anterior fue debido a la persistencia del tumor residual hipofisiario y la dificultad en el control bioquímico de su enfermedad, junto la imposibilidad de tratarla con terapias complementarias como pegvisomant que no estaban disponibles en esa época en el país.
En la actualidad recibe monoterapia con una dosis de 120 mg mensuales de Lanreotide, logrando que la somatomedina C esté en metas (Tabla 1). En la última resonancia de silla turca disponible hay una lesión residual que se proyecta hacia el seno esfenoidal izquierdo, pero que ha disminuido de tamaño según los estudios previos. Las manifestaciones derivadas de la acromegalia han desaparecido y tampoco ha tenido signos de hipercortisolismo. No se ha documentado recurrencia del schwannoma ni la presencia de mixomas cardiacos. Tres años después del inicio del Lanreotide requirió de colecistectomía por aparición de colelitiasis.
No hay evidencia de afectación familiar con esta misma condición. Se realizó un estudio genético que reportó una mutación en el gen PRKAR1A: c.682C > T(R228X), localizada en el exón 7, en la que hay una sustitución de una sola base. Esta mutación ya ha sido descrita previamente en la literatura (5).
Discusión
El complejo de Carney (CNC) es un síndrome raro. De herencia autosómica dominante, descrito en 1985 por J. Aidan Carney (1) como la combinación de lentiginosis, endocrinopatía y mixomas atriales. Hay una asociación entre la neoplasia endocrina múltiple, las manifestaciones cardio-cutáneas y los tumores no endocrinos. Con una edad promedio en el diagnóstico de 20 años (2,6-8) y un predominio en las mujeres (63 % de los casos) (6,7).
Las alteraciones en la pigmentación cutánea están presentes desde el nacimiento. La lentiginosis se presenta en la mayoría de los pacientes (70 %) como máculas hiperpigmentadas alrededor de los labios, conjuntivas, cantos de los ojos y mucosa genital. Toma su distribución característica cerca de la pubertad (6,7,9,10) y tiende a desaparecer hacia la cuarta década (Figura 2) (2,6). También se pueden observar nevus azules, manchas café con leche y mixomas cutáneos hasta en el 20 % de los casos, los cuales son pápulas sésiles o pediculadas localizadas en los párpados, lengua, paladar, pabellón auricular, pezones, glúteos y periné (6,7,10-12). Se han descrito los acrónimos LAMB (léntigos, mixomas auriculares, nevus azules) y NAME (nevus, mixomas auriculares, neurofibromas mixoides y efélides) para describir las manifestaciones cutáneas del síndrome (10). La presencia de mixomas cutáneos se puede asociar con mixomas cardiacos hasta en el 80 % de los casos (13), situación que no se presentó en nuestra paciente.
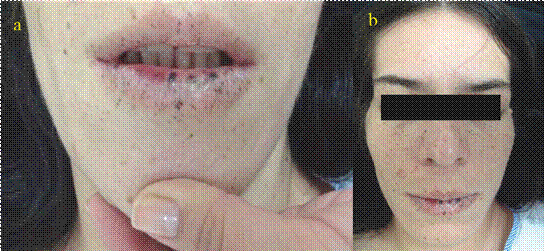
Fuente: creación propia
Figura 2 Características fenotípicas del complejo de Carney. a. Lesiones lentiginosas en mucosas características del CC. b. Cambios faciales por acromegalia.
Los mixomas cardiacos se encuentran hasta en el 60 % de los casos (8,14). Son la segunda manifestación más frecuente del síndrome después de los léntigos y se encuentran a una edad más temprana que los mixomas esporádicos (13,15). No tienen predilección por sexo y se pueden formar en cualquiera de las cavidades cardiacas, aunque son más prevalentes en la aurícula izquierda (75 % de los casos) (13,15-18). Su tamaño varía desde 2 mm hasta 15 cm (18); pueden ser múltiples, recurrentes y estar ubicados en varias cavidades al tiempo. Son causa de mortalidad específica hasta en un 50 % de los afectados, se asocian con una enfermedad cerebrovascular embólica, embolismo pulmonar, arritmias, angina de pecho, infarto del miocardio, obstrucción del flujo cardiaco por protrusión a través de las válvulas con muerte súbita o insuficiencia cardíaca (6-8,13-19). Las personas con CC y mixomas tienen un riesgo 5 veces mayor de presentar embolismos en comparación con aquellos con mixomas no asociados al CC (20). También se han descrito mixomas intravasculares (por ejemplo, los intracarotídeos) (21). Algunos pacientes pueden tener síntomas constitucionales debido a la producción de interleuquina 6 por el tumor (18). Para el diagnóstico se puede usar la ecocardiografía, tomografía o resonancia magnética (15,18,19). El manejo consiste en la extirpación quirúrgica de las lesiones seguido de vigilancia ecocardiográfica, pues se tiene un mayor riesgo de recurrencia (22 %) comparado con los casos de mixomas no asociados al CC (15,16,19). Además, se ha utilizado como alternativa en un caso la extracción por vía percutánea del tumor con el uso de radiofrecuencia, monitoreo con ecocardiografía intracardiaca y protección para embolismo, con buen resultado (19). En pacientes muy jóvenes con adenoma hipofisiario sometidos a cirugía de mixomas, se debe considerar la posibilidad de la apoplejía hipofisiaria por sangrado intratumoral asociado con la anticoagulación, lo cual ya ha sido descrito en la literatura (22).
Dentro de las manifestaciones endocrinas más frecuentes del CC está el síndrome de Cushing de origen adrenal (independiente de ACTH), secundario a una hiperplasia adrenal bilateral micronodular; esta se divide a su vez en dos entidades: la enfermedad adrenocortical nodular pigmentaria primaria (EANPP) y la enfermedad adrenocortical nodular aislada (23). La EANPP se presenta en un 20-60 % de los pacientes con CC (6,7,24). Sin embargo, esta condición se ha encontrado en casi todos los pacientes con CC que han sido llevados a autopsia (2,6,23). Además de la hiperplasia, se han descrito casos de adenoma y carcinoma adrenocortical (23,25) y también, aunque de forma muy infrecuente, se han descrito casos de enfermedad de Cushing por adenomas hipofisiarios (26). Los pacientes pueden tener una forma clásica del síndrome de Cushing, pero también formas subclínicas (19 %) o cíclicas (14 %) (6,7,12,23,24,27). Se presenta principalmente en niños y en mujeres, puede aparecer antes de los 5 años, pero también lo puede hacer en adultos jóvenes. Se ha descrito que 1 de cada 3 pacientes puede no tener los hallazgos característicos en la tomografía de adrenales como ocurrió en nuestro caso, o presentarlos sin nunca desarrollar el síndrome de Cushing (6,7,13,23,24,27,28). Un hallazgo diagnóstico importante es el aumento paradójico del cortisol urinario (mayor al 50 %) durante el segundo día tras recibir dexametasona en el test de Liddle, o el incremento del cortisol sérico tras la administración de 1 mg de dexametasona a las 11 p.m, asociado con la supresión de la hormona adrenocorticotropa (ACTH) (5,12,13,29).
En el 72 % de los casos los pacientes con esta condición tendrán también una acromegalia (30), como se presentó en nuestra paciente. El manejo consiste en la adrenalectomía bilateral, aunque también se ha descrito la adrenalectomía unilateral, sin evidencia de hipercortisolismo posterior en un seguimiento por más de 10 años (24). En nuestra paciente se hizo una adrenalectomía unilateral ante el hallazgo por imágenes de un adenoma, tras la cual hizo una fase de insuficiencia adrenal y, posteriormente, una recuperación de la función adrenal sin evidencia posterior clínica o bioquímica de hipercortisolismo (24). Se han descrito casos de remisión espontánea del hipercortisolismo en pacientes que no fueron llevados a cirugía (6,24). Como alternativa a la cirugía está el manejo médico con ketoconazol o mitotane (6,29).
Los adenomas secretores de hormona de crecimiento (GH) se reportan en el 12 % de los casos del CC (7,8,12,31,32), presentándose entre la tercera y cuarta década (2,6,7), con una edad promedio de 28,8 ± 12 años en un reporte de 57 casos de pacientes con CC, una edad más temprana a la de los casos esporádicos de acromegalia que se encuentran entre los 40 a 48 años (32). Sin embargo, hasta en un 75 % de los pacientes se pueden encontrar elevaciones sutiles y asintomáticas de la GH e IGF-1, antes de que sea evidente un adenoma en los estudios imaginológicos debido a una hiperplasia de somatótropos (6,7,12,31-37).
Se ha reportado que, en muchos casos, los valores de GH e IGF-1 no son tan altos como en la acromegalia no sindromática (32). También se ha descrito la presencia de múltiples adenomas en estos pacientes (6,7,38). Hay una proporción similar de macro y microadenomas (32), aunque otros autores sugieren el predominio de macroadenomas (6,30,36). En la mayoría de los casos en el momento del diagnóstico de la acromegalia ya están presentes otros signos del CC (36,38,39). No es infrecuente encontrar adenomas mixtos con prolactina y, en un caso, se describió un adenoma cromófobo asociado con acromegalia; otros 2 casos de tumores corticotropos y uno de tirotropo (6,31,38,40). Los prolactinomas aislados son muy raros (6).
Se ha descrito que los pacientes con CC que tienen acromegalia o elevaciones de la GH/IGF-1 tienen un riesgo 2,7 veces mayor de tener mixomas cardiacos durante su vida (41). En nuestra paciente ha sido llamativo la ausencia de mixomas cardiacos durante su evolución a pesar de la acromegalia de base y de tener mixomas cutáneos.
En la mayoría de los casos el manejo es quirúrgico, aunque también se ha reportado tratamiento con bromocriptina, agonistas de somatostatina, antagonistas de somatropina o radioterapia (6,7,32,41). En los casos de adenomas múltiples se puede hacer resección de estos o hemihipofisectomía (6,7,38). Estos pacientes pueden tener una menor tasa de éxito en el control hormonal con la cirugía transesfenoidal, debido a la hiperplasia de somatótropos que se pueden presentar acompañando al adenoma (42). En nuestro caso se realizaron dos cirugías transesfenoidales con resección incompleta del adenoma; se inició el manejo farmacológico ante la persistencia de la sintomatología y los valores altos de las hormonas (Tabla 1). Se requirieron dosis altas de análogos de somatostatina en combinación con agonista dopaminérgico. No se consideró el manejo con radioterapia por la edad de la paciente y el riesgo de inducir un hipopituitarismo. Con el paso de los años se lograron normalizar los niveles de somatomedina C, aunque la somatropina ha estado ligeramente por encima de las metas. Se logró el control sintomático, el desmonte del agonista dopaminérgico y se disminuyó la dosis de Lanreotide a 120 mg al mes. Como complicación de la terapia se presentó una colelitiasis que requirió de manejo quirúrgico.
En el CC se describen de forma menos frecuentes los quistes, adenomas y carcinomas tiroideos (folicular, papilar). A diferencia de la población general donde el carcinoma papilar es el más común. En el CC el carcinoma folicular es el más frecuente, se caracteriza por ser multifocal y bilateral con un mayor riesgo de presentar compromiso ganglionar en el cuello, metástasis y mayor mortalidad respecto a la variante no sindromática, principalmente, cuando tiene un tamaño superior a 3 cm (43). Además, se ha reportado hipertiroidismo por enfermedad de Graves, adenoma tóxico o bocio multinodular (6,7,43).
En hombres se presentan con frecuencia tumores testiculares (41-50 % de los pacientes), específicamente, el tumor de células de Sertoli grande calcificante, que puede ser multifocal, bilateral y funcional, causando ginecomastia y pubertad precoz (6,7,25,44), debido a la producción de aromatasa (45). Se identifican como masas palpables y en la ecografía son características las calcificaciones y la imagen en árbol de navidad (6,7,25,46). Se puede asociar con malignidad, principalmente cuando se presenta en la cuarta década de la vida. En adultos puede causar infertilidad debido a la compresión de los conductos seminíferos o a las alteraciones en la producción hormonal (45). También se han descrito tumores de células de Leydig y restos de adrenales gonadales, que podrían producir un síndrome de Cushing por EANPP (13). El manejo es quirúrgico, pero también se ha descrito con inhibidores de la aromatasa para tratar la pubertad precoz y la ginecomastia (44,46).
En las mujeres son comunes los quistes foliculares ováricos (14-66 % de las pacientes) (6,7,47), que pueden ser múltiples y complejos (48). Se han descrito también los teratomas, el adenocarcinoma mucinoso metastásico, el adenocarcinoma adenomatoide y tumores mixoides uterinos (7,48). Manifestaciones más inusuales incluyen mixomas o adenomas ductales de mama y los angiomixomas (6,7,12,14).
El schwannoma melanocítico psamomatoso (SMP) se presenta hasta en un 18 % de los casos (8), puede aparecer tanto en el sistema nervioso central como en el periférico. Sin embargo, es más frecuente en el tracto gastrointestinal y en la cadena simpática paravertebral o en la pared torácica y, ocasionalmente, puede comprometer los tejidos blandos, la piel, el hígado o el corazón (49-51). Suele aparecer alrededor de los 22 años (49,50). Histológicamente, estos tumores presentan depósitos de melanina, cuerpos de psammoma y calcificaciones (6,9). Pueden ser multifocales, lo que en muchas ocasiones dificulta el diagnóstico diferencial con metástasis de una lesión primaria. Pueden recurrir en el 35 % de los casos y tener metástasis hasta en el 44 % (50). Tras la resección de la lesión torácica nuestra paciente no ha tenido recurrencia de esta neoplasia.
La malignidad en el CC se observa principalmente en los nódulos tiroideos y en el SMP (7,8), pero también en los tumores testiculares y ováricos. En el 25 % de los casos de CC la muerte se debe a la enfermedad metastásica tumoral (46).
Otras manifestaciones que también se han descrito en el CC son los adenomas hepatocelulares, carcinoma de células acinares, adenocarcinoma pancreático, neoplasia intraductal mucinosa del páncreas, osteocondromixomas, tumor mixto de parótida, quistes broncogénicos, carcinoma gástrico o del colon e histiocitomas fibrosos peritoneales (7).
Se han descrito varios genes involucrados en la fisiopatología del CC, de estos el más importante es el CNC1, localizado en 17q22-24. También está el gen CNC 2 localizado en 2p16. El CNC1 es un supresor tumoral que codifica la subunidad regulatoria R1A de la proteína quinasa A (PRKAR1A). Este gen tiene 11 exones, de los cuales del 2 al 11 tienen la función de inducir la síntesis de proteínas (12). PRKAR1A es un componente clave en la vía de señalización de la adenosina monofosfato cíclica (AMPc), que actúa sobre la subunidad alfa de la proteína G de los receptores de la membrana. La proteína quinasa A de serina-treonina dependiente de AMPc participa en un gran número de procesos celulares como la transcripción, el metabolismo, la progresión en el ciclo celular y la apoptosis (3,6,7). Las mutaciones con pérdida de función, la sustitución de bases, inserciones, re-arreglos o las deleciones del PRKAR1A, de las cuales hay más de 140 descritas en la actualidad, van a llevar a un aumento en la señalización del AMPc celular, con activación de la vía AMPc/proteína quinasa A, y la consiguiente activación de las mitosis, proliferación celular y la aparición de las diferentes manifestaciones del síndrome. Varias mutaciones heterocigóticas inactivantes, que generan un codón de parada, están involucradas en la hiperactividad endocrina (3,6,7,9,12,13, 52 ,53). Contrario a los casos de CC sin mutaciones del PRKAR1A, los que las presentan tienen una mayor frecuencia de mixomas, lesiones de la piel, schwannomas, tumores tiroideos y gonadales, con una edad de aparición más temprana (13,53). En algunos individuos con criterios para CC que no presentan mutaciones del PRKAR1A se han encontrado alteraciones en un locus en el cromosoma 2p16, conocido como CNC2. El gen no está claramente identificado, pero se considera que está involucrado en la misma vía molecular del PRKAR1A ante la similitud del compromiso clínico y fenotípico (3).
También se han descrito otras afectaciones genéticas que pueden causar el CC. Se han definido alteraciones en el gen de la subunidad catalítica C alfa del complejo proteína quinasa A (PRKACA), y de la subunidad catalítica beta del complejo de proteína quinasa A (PRKACB) (3). Las variantes activantes de la PRKACA se asocian con el síndrome de Cushing, EANPP y a adenomas productores de cortisol (3). En el caso de la PKKACB, su sobre expresión se asocia con la acromegalia, lesiones pigmentadas cutáneas y mixomas en ausencia de síndrome de Cushing. Todos se relacionan con la vía de señalización del AMPc (3).
Cuando se documenta una mutación en PRKAR1A, como en nuestra paciente, se debe realizar un análisis genético a todos los familiares en primer grado (11). En su familia no hay ningún familiar que tenga antecedente de CC. A la fecha, a ninguno de ellos se les ha podido realizar el estudio genético en sus aseguradoras. Podría ser un caso esporádico con una mutación de novo, pues sus familiares son adultos y no han desarrollado manifestaciones del síndrome que deberían haber aparecido a temprana edad.
El 50-70 % de los casos de CC de origen familiar van a tener la mutación (2,3,6,7), mientras que en los esporádicos el 37 % (3,7). Detectar estas mutaciones es de vital importancia para realizar una consejería genética, pues la penetrancia de la enfermedad es muy alta, cercana al 95 % a los 50 años (3,7,32).
Hoy el diagnóstico de CC se hace cuando 2 o más de las manifestaciones principales están presentes, confirmadas por histología, análisis bioquímico o por imágenes. También cuando el paciente tiene una manifestación asociada y un familiar en primer grado con el síndrome o si tiene una mutación del PRKAR1A (Tabla 2) (3,7,9).
Tabla 2 Criterios diagnósticos del CC
| Criterios diagnósticos |
|---|
| Lesiones cutáneas pigmentadas con una distribución típica (labios, conjuntiva, canto interno y externo, mucosa vaginal y del pene) * |
| Mixoma (cutáneos y de mucosas) * |
| Mixoma cardiaco |
| Mixomatosis de la mama o hallazgos sugestivos en una resonancia con supresión de grasa |
| EANPP o respuesta paradójica del cortisol urinario después de una supresión con dexametasona en un test de Liddle* |
| Acromegalia por un adenoma productor de GH* |
| Tumores de Sertoli o calcificaciones características en ecografía testicular |
| Carcinoma de tiroides o múltiples nódulos hipoecoicos en la ecografía en un paciente joven |
| Schwannoma melanocítico Psammomatoso* |
| Nevus azules y nevus azules epitelioides (múltiples) |
| Adenoma ductal de mama (múltiple) |
| Osteocondromixoma |
| Criterios suplementarios |
| Familiar en primer grado afectado |
| Mutación inactivante del gen PRKAR1A* |
El diagnóstico se hace con 2 de las manifestaciones clínicas o con 1 de las manifestaciones más 1 de los criterios suplementarios. * Criterios que presentó la paciente. Fuente: adaptado de (3,7,9)
Recomendaciones
Los pacientes con CC o mutación PRKAR1A deben tener un seguimiento anual de las manifestaciones de la enfermedad; el tratamiento es específico para cada tumor, siendo los schwannomas los más difíciles de tratar, considerando que, frecuentemente, son inoperables y metastásicos (9). Se debe hacer seguimiento ecocardiográfico anual desde la infancia, o semestral si se requirió de la resección de un mixoma (7,46,53). En los niños se debe hacer vigilancia de la velocidad de crecimiento y el desarrollo sexual en la etapa prepuberal; eco pélvica y mamaria en las mujeres y eco testicular en hombres desde la pubertad con control anual si se identifica alguna alteración. Además, ultrasonido tiroideo y monitorización anual de los niveles de IGF-1, prolactina, TSH, tiroxina libre, ACTH y cortisol urinario o cortisol posdexametasona desde la adolescencia, y la realización de imágenesy test diagnósticos específicos si hay sospecha de un tumor funcional, como es el caso de la resonancia magnética de silla turca y tomografía, resonancia de tórax, abdominal o pruebas dinámicas de laboratorio (6,7,46,53). El manejo de la acromegalia y del síndrome de Cushing es quirúrgico, dejando como alternativa el tratamiento farmacológico para aquellos casos en los que no se logró un control adecuado con la resección tumoral.

