










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Universidad Industrial de Santander. Salud
Print version ISSN 0121-0807On-line version ISSN 2145-8464
Rev. Univ. Ind. Santander. Salud vol.41 no.2 Bucaramanga May/Aug. 2009
Leucemia mieloide crónica,
estado del arte
Luz Aída Rey1; Yenny M. Montenegro2
1. Bacterióloga. Universidad Industrial de Santander, Email:
2. Bacterióloga MSc Ciencias Básicas Biomédicas. Docente Escuela de Bacteriología, Universidad Industrial de Santander.
Correspondencia:
Yenny M. Montenegro Medina. Bacterióloga Msc Ciencias Básicas Biomédicas. Docente Escuela de Bacteriología. Universidad
Industrial De Santander. Email:yenny_mmm@yahoo.com Tel: 6384000 Ext: 3135.
Recibido: 20 enero de 2009 -Aceptado: 11 de marzo de 2009
RESUMEN
La leucemia mieloide crónica es un desorden clonal maligno producido por la translocación genética t(9;22)(q34;q11), que genera hiperplasia en médula ósea y proliferación incontrolada de la línea mieloide en sangre periférica. Hasta 1980, se consideró una enfermedad fatal, sin embargo gracias a los avances científicos se empezaron a dilucidar diferentes estrategias terapéuticas, que han ido desde el trasplante alogénico de médula ósea, hasta el desarrollo de fármacos de última generación como es el caso de los inhibidores tirosin kinasa de primera y segunda generación (Imanitib y Nilotinib), con los cuales se ha obtenido una respuesta positiva hasta en un 95% de los casos que ha obligado a nuevas estrategias diagnósticas y de seguimiento como la fluorescencia por hibridación in situ y las diferentes variantes de la reacción en cadena de la polimerasa; conocer estos avances es fundamental para nuestro desempeños como profesionales de la salud, ya que nos permite actuar y tomar decisiones acertadas para el beneficio del paciente, acordes con los recursos del medio. Salud UIS 2009; 41: 169-180
Palabras Clave: Leucemia mieloide crónica BCR-ABL positiva, translocación genética, síndrome mieloproliferativo, imatinib
Myeloid cronic leukemia, state of the art
ABSTRACT
Myeloid chronic leukemia is a malignant clonal disorder caused by genetic the translocation t(9; 22) (q34, q11) which generates hyperplasia in bone marrow and uncontrolled myeloid proliferation in peripheral blood. Until 1980, it was considered a fatal disease, however, thanks to scientific progress, scientists began to elucidate different therapeutic strategies, from allogeneic transplantation of bone marrow, to the first and second generation tyrosine kinase inhibitors (Imanitib and Nilotinib), which has provided a positive response in up to 95% of cases. Such development has enforced new diagnostic and monitoring strategies as the fluorescence in situ hybridization and different variants of the polymerase chain reaction. Knowing such advances is fundamental to our performance as health care professionals, allowing us to act and make sound decisions for the benefit of the patient, according to available resources. Salud UIS 2009; 41: 169-180
Keywords: Leukemia myelogenous chronic, BCR-ABL positive, translocation genetic, myeloproliferative syndromes, imatinib
INTRODUCCIÓN
La leucemia mieloide crónica (LMC) es un síndrome mieloproliferativo crónico (SMPC) de naturaleza clonal y con origen en una célula madre pluripotencial (CMP) común a tres series hematopoyéticas. Los síntomas más comunes son: fatiga, anorexia y pérdida de peso, pero entre el 40 y 50% de los pacientes son asintomáticos al momento del diagnóstico1, 2.
La LMC, representa entre el 15 y el 20% del total de las leucemias diagnosticadas y su incidencia en Europa y Estados Unidos es de 1 a 2 casos/100,000 habitantes/ año. Predomina ligeramente en varones (1,3 hombres por cada mujer) y la edad promedio de aparición es de 53 años. Entre el 12 y el 30% son mayores de 59 años y aproximadamente el 10% de los casos se presenta en niños y adolescentes menores de 20 (Figura 1)1-3. En Colombia, se informan entre 400 y 800 casos anualmente y existen a la fecha entre 2000 y 3200 pacientes con la enfermedad3, pero no existen datos precisos; en Santander, de acuerdo con el Observatorio de Salud Pública, a pesar de no tener datos específicos sobre LMC, se logró establecer que se diagnosticaron más de 420 casos de leucemia en los servicios de consulta sólo en el 2005, con una incidencia en Bucaramanga y el área metropolitana de 11,3 casos/100,000 habitantes/ año3. Si asumimos un porcentaje de diagnóstico similar al mundial y estimamos la LMC como el 20% de los casos en nuestra población, podríamos decir que en Santander durante el 2005 pudieron haberse presentado 84 casos de LMC (2,26 casos/100,000 habitantes/año).
La LMC fue la primera enfermedad humana asociada a una anormalidad citogenética específica "El Cromosoma Filadelfia (Ph)", presente en más del 95% de los casos y directamente relacionado con la iniciación, progresión, manifestaciones clínicas y terapéutica de la enfermedad. Este cromosoma ha sido también identificado en el 5% de los niños con leucemia linfoide aguda (LLA) y en el 15 a 30% de los adultos con esta misma enfermedad; también se encuentra presente en el 2% de los casos de leucemia mieloide aguda (LMA)1-5. Es importante mencionar que a medida que progresa la enfermedad, las células oncogénicas son genéticamente más inestables, lo cual, acompañado de la alta tasa de proliferación conduce a la acumulación de otras anormalidades cromosómicas (Tabla 1). A este proceso se le conoce como evolución clonal (EC) y es considerado de mal pronóstico, ya que afecta la diferenciación celular y conduce a la transformación de 2/3 de los casos de LMC como LMA y los restantes como LLA1-5.
Etiología de la enfermedad
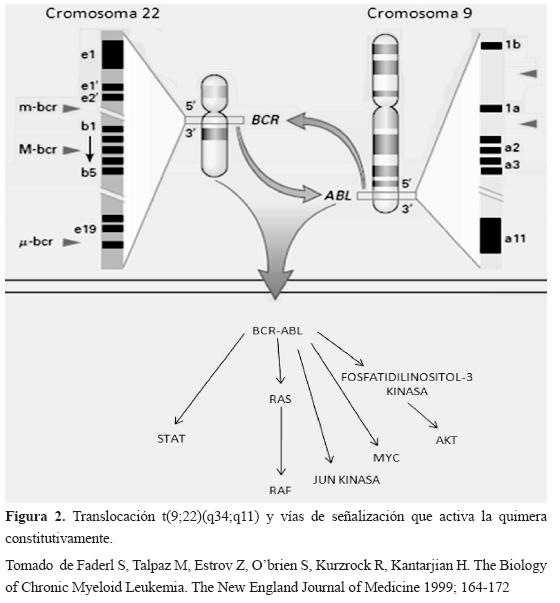
El cromosoma Ph resulta de la translocación reciproca entre los brazos largos de los cromosomas 9 y 22, t(9;22)(q34;q11) (Figura 2), implicando el traslado del segmento 3' del protooncogen abl (Abelson) desde cromosoma 9q34 hasta el extremo 5' del gen bcr (breackpoint cluster región) sobre el cromosoma 22q113-5, creando un gen quimérico de fusión bcr-abl con actividad tirosin quinasa constitutiva (activo constantemente)3-5y confinada al citoplasma; mientras que el gen normal se encuentra migrando entre el núcleo donde tiene actividad antiapoptotica y el citoplasma donde su función es fosforilar sustratos como: CRKL (oncogen homologo de la proteína V-crk del virus CT10 del sarcoma aviar), p62Dok (Proteína de unión 1, 62kDa, de la tirosin quinasa (1), conocida también como DOK1, CBL (oncogen de transformación antirretroviral murino, CAS-Br-M) y RIN (Proteína efectora de RAS, interacción/interferencia) y por tanto, activar múltiples vías de señalización que intervienen en el crecimiento, adhesión y la diferenciación celular de la línea hematopoyética como: RAS (sarcoma de ratón), RAF ( Proto-oncogen serina/treonina kinasa), AP1 kinasa (factor de trascripción 1), IP3 kinasa (inositol trifosfato kinasa), MYC (Mielocitomatosis) y STAT (traductor de señales y activador de la trascripción)1-10 (Figura 2).
Dependiendo del punto de ruptura en el gen bcr, la proteína de fusión BCR-ABL puede variar de tamaño pues la ruptura en abl es muy variable, pero siempre se da corriente arriba del exón a2 (Figura 3). Hasta la fecha, han sido identificados 3 puntos de ruptura en bcr denominados: M-BCR (Región Mayor), m-BCR (Región Menor) y m-BCR (Región Micro). La ruptura de mayor frecuencia es M-BCR sobre el intrón ubicado entre los exones e13 y e14 (anteriormente exones b2 y b3) o en el intrón entre los exones e14 y e15 (anteriormente b3 y b4) generando una proteína BCR-ABL de 210Kd (p210BCR-ABL); los dos puntos de ruptura dan como resultado quimeras ligeramente diferentes, una e13a2 (ó b2a2) y otra e1a2 (ó b3a2), generalmente se presenta una de las dos; sin embrago aproximadamente el 10% de los pacientes son heterocigotos. Cuando la ruptura es sobre el primer intrón de bcr (m-BCR) entre los exones e1 y e2, se transcribe una proteína de 190Kd (p190BCR-ABL), la cual está presente en aproximadamente 2/3 de los pacientes con LLA Ph+. El tercer punto de ruptura ocurre entre los exones e19 y e20 (m-BCR), el ARN producido por esta quimera codifica la proteína más larga de las tres, con un tamaño de 230Kd (p230BCR-ABL) común en leucemia neutrofílica crónica asociada a Ph+ 1-12, 18 (Figura 3).
Historia natural
La historia natural de la LMC está dada por una continua progresión desde una fase crónica donde el recuento de leucocitos es alto pero la maduración es normal, hasta una fase blástica caracterizada por el aumento de blastos en médula ósea (MO) y sangre periférica (SP) superior al 20% acompañada de defectos en la maduración en un periodo no superior a 5 años cuando no se recibe tratamiento oportuno. La crisis blástica generalmente se encuentra precedida por una fase de aceleración que se evidencia por una neutrofília marcada y otros signos, tal y como se describe en la (Tabla 2). 1-5,11-12
Diagnostico
El 80% de los pacientes es diagnosticado en fase crónica1. Los signos y síntomas que se presentan comúnmente son: fatiga(80%), astenia(51%), anorexia(37%), pérdida de peso(36%), dolor abdominal y/o saciedad precoz(34%), esplenomegalia(40%) y dolor óseo(10%); aunque en el 50% de los casos no se manifiesta sintomatología de importancia al diagnóstico y éste se realiza de forma accidental con el hemograma al hallar: anemia normocítica normocromica, leucocitosis (>25.000 células/mm3), desviación a la izquierda de la línea mieloide sin hiato leucémico, linfopenia, basofilia ocasional y, trombocitosis (30-50%)2-5.
Al estudio de MO, esta se encuentra hipercelular y similar a SP. Con frecuencia hay megacariocitosis y la relación mieloide/eritroide (M:E) está elevada. No obstante, la confirmación del diagnostico se hace con la identificación del cromosoma Philadelfia (Ph) y/o la detección de transcritos BCR-ABL por Reacción en Cadena de la Polimerasa (PCR)11-17.
El cariotipo convencional, es hasta ahora la técnica más empleada para el diagnóstico confirmatorio de la LMC, a pesar de la poca sensibilidad y el advenimiento de la biología molecular; ya que es la única prueba capaz de detectar la EC11, 14-17.
La fluorescencia por hibridación in situ (FISH), identifica "microarreglos simples o complejos como "microPh" no detectables por técnicas convencionales de citogenética"; puede realizarse en diferentes sustratos (sangre, médula ósea, fluidos corporales, tejidos fijados con parafina) y, tiene una especificidad (98%) y sensibilidad (60%) superior al cariotipo, pero no detecta EC y es costosa. Se usa cuando el cariotipo convencional es negativo y en algunos casos como estrategia de seguimiento11,13.
La PCR, detecta una célula BCR-ABL (+) entre 105 y 106 normales, convirtiéndose en la herramienta más sensible hasta la fecha y la técnica de elección para el diagnóstico en pacientes Ph(-). La PCR en tiempo real, es una modificación de la PCR convencional que permite cuantificar el número de trascriptos BCR-ABL circulantes en el paciente; por eso aunque un poco más complejo, es el método más preciso para llevar a cabo el seguimiento y monitoreo de los pacientes con LMC que están en tratamiento. En Colombia, esta metodología ya ha sido implementada en ciudades como Bogotá y Medellín11, 15.
Seguimiento
El objetivo del seguimiento es identificar cualquier resistencia al tratamiento en forma temprana, permitiendo diseñar estrategias terapéuticas alternativas encaminadas a retardar la progresión de la enfermedad. Esta respuesta es medida mediante la evaluación de tres tipos de respuestas: la hematológica, citogenética y molecular (Figura 4).
Respuesta hematológica (RH): Se habla de RH, cuando el hemograma empieza a normalizarse (disminución progresiva de los recuentos leucocitarios). Sin embargo, para hablar de respuesta hematológica completa (RHC), es indispensable que exista un recuento de plaquetas > a 450.000/mm3, leucocitos < a 10.000/mm3, 0% de células inmaduras en SP, basófilos < al 5% y normalización del tamaño del bazo. Se calcula que un paciente en el momento de su diagnóstico tiene una carga tumoral de 1012 células leucémicas en su cuerpo, al obtenerse la RHC esta carga se reduce a 1011 células, siendo éste su límite de detección. El monitoreo hematológico debe realizarse cada dos semanas hasta evidenciar la RHC, y posterior a ella cada 3 meses1-5,11-23.
Respuesta citogenética (RC): Está determinada por el porcentaje residual de células Ph(+)en metafase, clasificándose como: Ninguna si existen más del 95% de células Ph(+), Mínima de 66-95%, Menor (RCm) de 36-65%, Mayor (RCM) de 1-35% y Completa (RCC) cuando no se detectan células Ph(+). El cariotipo se considera la metodología de elección para este seguimiento, pues no solo determina la presencia del cromosoma Ph, sino que también facilita la detección de EC. Debe realizarse cada 6 meses11-19.
Se espera que en un paciente que responde óptimamente al tratamiento, la RHC deba generarse 3 meses después de iniciado el tratamiento, la RCm a los 6 meses, la RCM en el transcurso de un año y en máximo 18 meses, se debe lograr una RCC. Cualquier incumplimiento de estos criterios debe llevar a la reevaluación y planteamiento de nuevas estrategias terapéuticas11-12, 19-22.
Un pequeño porcentaje de pacientes que alcanzan RCC recaen. La recaída surge de la persistencia de células malignas por debajo del nivel de detección por citogenética; este reservorio se conoce como enfermedad mínima residual (EMR), la cual corresponde a una carga tumoral por debajo de 109 a 1010 células leucémicas, y puede ser detectada sólo por métodos moleculares. Es por esto que el concepto de respuesta molecular debió ser incluido1-5, 19-23.
Respuesta molecular (RM): Se conoce como RM, la proporción del gen BCR-ABL, del transcrito o de la proteína quimérica, dependiendo del método de detección, presente en los pacientes con LMC. Existen dos tipos de RM, una respuesta molecular mayor (RMM) que se define como la reducción en mínimo 3 log de los transcritos BCR-ABL y una respuesta molecular completa (RMC), entendida como la no detección del transcritos BCR-ABL en MO y SP11-15, 22.
Tratamiento
Hasta 1980, la LMC se consideró una enfermedad fatal, sin embargo desde su descripción hasta nuestros tiempos, nuevas estrategias terapéuticas han surgido para su tratamiento; cada una buscando superar las debilidades de la otra. A continuación, haremos una breve presentación de forma cronológica de los principales tratamientos hasta la fecha desarrollados para el tratamiento de la LMC.
Busulfan (1,4- Butanodiol Dimetilsulfonato): Conocido comercialmente desde 1959 como Myleran (GlaxoSmithKline). Es un agente alkilante, específicamente un alfil sulfonato que se caracteriza por su capacidad de formar uniones covalentes con sustancias nucleofílicas y producir iones carbonilo, los cuales reaccionan instantáneamente con aminas, grupos oxhidrilos y sulfhidrilos, interrumpiendo la síntesis del ADN y la división celular, por lo que se le adjudica un efecto inmunosupresor, ya que disminuye los recuentos de leucocitos, controlando las manifestaciones hematológicas de la enfermedad (en el 90% de los casos). Por tanto, mejora la calidad de vida, pero sin retrasar la progresión de la enfermedad. Tiene cierto grado de toxicidad, llegando a generar fibrosis pulmonar19-22.
Hidroxiuria (CH4n202) o hidroxicarbamida: es un antimetabòlico análogo de la úrea conocido comercialmente como Droxia o Hydrea. Inhibe la ribonucleótido-reductasa, enzima que transforma los ribonucleótidos en desoxirribonucleótidos; por lo cual impide la síntesis de ADN y con ello la replicación celular. Se desarrolló como una alternativa más segura en el tratamiento de la LMC debido a su menor toxicidad en comparación con el busulfan. Su uso se inicio en 1964, sin embargo ha sido remplazada, ya que al igual que el busulfan prolonga la supervivencia pero no impide la progresión de la enfermedad19-23.
Citarabina (4-amino-1-[(2R,3S,4R,5R)-3,4-dihidroxi-5-(hidroximetil)oxolan-2-yl] pirimidina): Fue descubierto en Europa en la década 1960 y aprobado por la Food and Drug Administration (FDA) en junio de 1969. También llamado arabinofuranosilcitosina, arabinosilcitosina, y Arabinósido de citosina o ara-C. Es agente antimetabólico análogo de la citosina que bloquea la síntesis y reparación del ADN por inhibición competitiva de ADN polimerasa24.
Trasplante alogénico de medula ósea (TAMO): El primero que se realizó con éxito en un paciente con LMC tuvo lugar en el año de 1970, y desde entonces ha evolucionado hasta ser el único tratamiento curativo al generar una RMC, alcanzando una supervivencia sin enfermedad de larga duración2-7,22. Para que el TAMO sea efectivo, es necesario que esté precedido por un tratamiento quimioterapéutico bastante agresivo que elimine el 100% de las clonas malignas y facilite la colonización de las células madres sanas. Este protocolo es altamente tóxico y mortal (10-70%); por lo que el TAMO se recomienda, en menores de 65 años o como segunda línea terapéutica tras el fracaso del imatinib25-27.
Debido a las restricciones de edad (relacionadas con las complicaciones), donantes compatibles y el riesgo del procedimiento, sólo del 15 al 20% de los pacientes con LMC son candidatos al TAMO y de estos; el 50% logra la RMC luego de 5 años; la cual, en algunos casos, se perpetúa, incluso hasta 20 años más, después del procedimiento. El 50% restante, generalmente recae por la permanencia de clonas malignas, que luego de algunos años inician nuevamente el proceso neoplásico28-31.
Interferón- alfa (INF-α): Es una glicoproteína de origen biológico que ha demostrado tener propiedades antivirales y antiproliferativa. Se introdujo como tratamiento de la LMC a principios de 1980 y en corto tiempo logro reemplazar la quimioterapia convencional, al demostrar mayor supervivencia de los pacientes (57% vs 42% en 5 años) y que era posible obtener la RCC en un pequeño porcentaje de pacientes sensibles al tratamiento (10-30%) sin inducir aplasia medular como ocurría con la quimioterapia intensiva. Al combinarse con quimioterapéuticos como la hidroxiurea o la citarabina en dosis bajas, puede llegar a alcanzarse en el 35 a 55% de los casos la RCC2-4, 41.
Poco se sabe sobre su mecanismo de acción en la LMC, pero se dice que aumenta la expresión del Antígeno Leucocitario Humano (HLA) en las clonas Ph(+), incrementando la presentación de antígenos a las células dendríticas. Este tratamiento sólo está indicado, en pacientes que se encuentran en fase crónica. El INF-α logró mantenerse como el tratamiento de elección hasta la aparición de los inhibidores específicos de la proteína Tirosina Kinasa y aún se considera un tratamiento válido en el manejo de la LMC28, 31-40.
Imatinib o Mesilato de Imatinib (STI 571, Gleevec): Es un activo con actividad antineoplásica no relacionado ni química ni farmacológicamente con otros medicamentos antineoplásicos42. Su aparición (1990) revolucionó por completo el tratamiento de la LMC llegando a ser aprobado en Europa y América en el 2001 como agente terapéutico de primera línea en esta patología, después de su éxito en numerosos ensayos clínicos. El Imatinib es un derivado de la fenilaminopirimidina (inhibidor de las proteínas tirosina kinasa) que compite con el Adenosin Trifosfato (ATP) por unirse al dominio SH1 de la proteína BCR-ABL, impidiendo que se lleve a cabo su fosforilación y por tanto, manteniéndola inactiva; de tal forma que inhibe selectivamente la proliferación de la clona neoplásica e induce la apoptosis de las mismas. Otra característica de gran importancia del imatinib es su alta especificidad por el dominio catalítico homologo del oncogen asociado al retrovirus originalmente aislado del sarcoma de Rous (Src) de la BCR-ABL. Aunque se ha visto, posee la capacidad de inhibir ciertas proteínas de señalización como el Receptor del Factor de Crecimiento Derivado de Plaquetas (PDGFR) y el c-Kit42-46.
Más del 90% de los pacientes en fase crónica tratados con Imatinib, han logrado RHC y entre el 70 y 80% de los casos también RCC. Sin embargo, cuando el tratamiento inicia en fase acelerada o en crisis blástica, la RHC disminuye al 76 y 34% respectivamente; y del mismo modo, del 76 al 91% no alcanzan la RCC47-48.
Actualmente, el Imatinib es el tratamiento de elección para el tratamiento de la LMC, pues ha demostrado que tanto la respuesta hematológica, citogenética y molecular, es muy superior a la obtenida con el IFN-α o los quimioterapéuticos convencionales, y adicionalmente, es muy bien tolerada clínicamente por la mayoría de los pacientes, desafortunadamente, cerca del 15-25% de los pacientes generan una resistencia primaria (intrínseca) al medicamento y un 20-30% de quienes responden inicialmente, luego de 5 años de tratamiento, adquieren una resistencia secundaria49-56.
Dasatinib (BMS-354825): Desarrollado por Bristol-Myers Squibb, actúa como un doble inhibidor de kinasas Src/Abl, es decir, inhibe a la familia de tirosina kinasas Src y además actúa como inhibidor selectivo de la kinasa de Abl. Es mucho más potente que el Imatinib (20-200 veces) en relación a su actividad sobre los mutantes y las formas salvajes del BCR-ABL, con la excepción del mutante T315I. Su unión con la proteína BCR-ABL, es más flexible, permitiéndole interactuar con diferentes estados conformacionales de BCR-ABL y por tanto, aumenta su afinidad. Aunque, el Dasatinib es el más potente inhibidor del dominio kinasa del ABL que se ha desarrollado hasta la fecha, no es el más específico, pues su finalidad es ampliar el perfil de reacción con el propósito de incluir a otros miembros de la familia Src7-68.
Nilotinib (AMN107): Derivado del Imatinib desarrollado por Novartis, actualmente en evaluación. Es un inhibidor selectivo del dominio kinasa del ABL en su estado inactivo. La estructura del Nilotinib le permite unirse más estrechamente a BCR-ABL aumentando su avidez, eficacia, especificidad y sensibilidad por la quimera neoplásica. Hasta el momento, la mayoría de pacientes resistentes a Imatinib son 25 a 50 veces más sensibles al Nilotinib (excepción la mutación T315I), lo cual lo hace mucho más potente que el Imatinib. El Nilotinib también inhibe al Receptor del Factor de Crecimiento Derivado de las Plaquetas (PDGFR) y el CD117 (receptor del factor de crecimiento de células Mast/stem), pero, a diferencia del Dasatinib, no inhibe la familia de kinasas Src. Hasta ahora este tratamiento se asocia con un perfil favorable de seguridad y tolerabilidad19,66-70.
Dos nuevos agentes, bosutinib (SKI-606) y INNO-406 (NS-187), han llegado a la Fase I - II de los ensayos clínicos. Sin embargo, ninguno de los agentes discutido hasta el momento es capaz de la inhibición de la mutante T315I de BCR-ABL.71, 72
Otras terapias innovadoras
Hommoharringtonina (HHT): Es una planta alcaloide derivada de un árbol chino. Su eficacia como agente anticancerígeno se ha reconocido y documentado tanto en la LMA como en la LMC. Se piensa que actúa inhibiendo la síntesis de proteínas. Se ha comprobado que su uso conjunto con el interferón alfa (IFN-α) o el Imatinib, potencia la respuesta al tratamiento, lográndose mejores resultados19,69-72.
Trióxido de arsénico (As2O3): No está claro cómo ejerce su acción en la LMC, pero se sugiere que posee capacidad apoptótica en las células malignas, reduciendo los niveles de BCR-ABL de forma especifica. Además, se ha descrito una relación sinérgica entre As2O3 y el Imatinib, incrementando la respuesta a estos fármacos71-73.
CONCLUSIONES
La LMC se caracteriza por ser una enfermedad con comportamiento bifásico o trifásico, diagnosticada en un 50% de los casos al momento del diagnóstico y su pronóstico depende de la etapa en la cual sea identificada, siendo la sobrevida mucho menor en los pacientes diagnosticados durante la fase acelerada o durante la crisis blástica.
Aunque el diagnóstico confirmatorio y pronóstico es definido con cariotipo y técnicas de biología molecular, no todos los pacientes tienen acceso a estas pruebas, por ser costosas y no estar incluidas en el Plan Obligatorio de Salud (POS), así como tampoco tienen acceso al uso de nuevos fármacos; disminuyendo la sobrevida de nuestros pacientes a menos de la mitad de la informada en países tercermundistas (5 a 7 años). Las principales razones por las cuales estos procedimientos diagnósticos y tratamientos no se encuentran cubiertos, puede deberse a la falta de gestión de los profesionales de la salud y el desconocimiento de las autoridades sanitarias sobre el número de casos y población que afecta (en su mayoría en edad productiva) y al desconocimiento de la evolución de las intervenciones terapéuticas durante la última década.
En Colombia, aunque el uso del imatinib fue aprobado desde enero de 2003 por el INVIMA como terapia de primera línea de la LMC y posteriormente recibió la aprobación para el uso en población pediátrica mayor de 3 años, no es un medicamento incluido en el POS, a pesar de que su uso es esencial para la vida y la salud del paciente. Esta situación es dramática si tenemos en cuenta que el beneficio del imatinib y otros inhibidores de las tirosina kinasas depende del cumplimiento disciplinado del tratamiento, sin la obstaculización a la que son objeto rutinariamente nuestros pacientes por diversas razones.
Finalmente, podemos decir que los espectaculares resultados con el imatinib, la existencia de dasatinib y nilotinib, la investigación activa de otra decena de compuestos potencialmente eficaces contra esta enfermedad, han hecho que el alotrasplante de médula ósea no se utilice hoy en pacientes con LMC en fase crónica como terapia de primera línea. Se puede decir que el advenimiento de los inhibidores de tirosin kinasas aumento el tiempo de supervivencia a más de cinco años en cerca de un 90% de los pacientes con LMC, cifra que en nuestro país con el trabajo de todos podría ser posible.
REFERENCIAS
1. Sessions J. Chronic Myeloid Leukemia in 2007. Journal of Managed Care Pharmacy 2007; Vol. 13: s4-s7 [ Links ]
2. Sawyers C. Chronic Myeloid Leukemia. The New England Journal of Medicine 1999; 1330-1340 [ Links ]
3. Rodríguez L, Hormiga C. Análisis de la Situación de las enfermedades neoplásicas en Santander. Observatorio de Salud Pública de Santander; 1-30. 2005 [ Links ]
4. Faderi S, Taplaz M, Estrov Z y cols. The Biology of Chronic Myeloid Leukemia. The New Engl J of Med. 341(3); 164-172. 1999 [ Links ]
5. Shirlyn B. Mckenzie. Hematología Clínica. 2da edición. Manual Moderno; 382-385. 2000. [ Links ]
6. Faderl S, Talpaz M, Estrov Z, O'brien S, Kurzrock R, Kantarjian H. The Biology of Chronic Myeloid Leukemia. The New England Journal of Medicine 1999; 164-172 [ Links ]
7. Carpino N, Wisniewski D, Strife A, et al. p62dok: A constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell 1997; 88: 197-204. [ Links ]
8. Salgia R, Uemura N, Okuda K, et al. CRKL links p210BCR-ABL with paxillin in chronic myelogenous leukemia cells. J Biol Chem 1995; 270:29145-50. [ Links ]
9. Afar DE, Han L, McLaughlin J, et al. Regulation of the oncogenic activity of BCR-ABL by a tightly bound substrate protein RIN1. Immunity 1997; 6: 773-82. [ Links ]
10. Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci U S A 1995; 92:11746-50. [ Links ]
11. Pavón V, Hernández P, Martínez G, et al. Leucemia Mieloide Crónica. Actualización en Citogenética y Biología Molecular. Rev Cubana Hematol Inmunol Med Transf 2006; 23(1). [ Links ]
12. Baccarani M, Pane F, Saglio G. Monitoring treatment of chronic myeloid leukemia. haematologica 2008; 93(2): 161 -166. [ Links ]
13. Mark Hon F, Sokolic R, Mark Y. Conventional cytogenetics and FISH in the detection of BCR-ABL fusion in chronic myeloid leukemia (CML). Experimental and Molecular Pathology 2006; 81:1-7 [ Links ]
14. Martiat P, Michaux JL, Rodhain J. Filadelfia-negative (Ph-) chronic myeloid leukemia (CML): comparison with Ph+ CML and chronic myelomonocytic leukemia. The Groupe Français de Cytogépeonétique Hématologique. Blood 1991; 78 (1): 205-11. [ Links ]
15. Gabert J, Beillard E, Vander V, Bi W, Grimwade D, Pallisgaard N, et al. Standardization and quality control studies of "real-time" quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - A Europe Against Cancer Program. Leukemia (2003) 17, 2318-2357 [ Links ]
16. Lundán T, Juvonen V, Mueller M, Mustjoki S, Lakkala T, Kairisto V, et al. Comparison of bone marrow high mitotic index metaphase fluorescence in situ hybridization to peripheral blood and bone marrow real time quantitative polymerase chain reaction on the International Scale for detecting residual disease in chronic myeloid leukemia. Haematologica 2008; 93(2): 178-185 [ Links ]
17. Mauro M, Deininger M. Chronic myeloid leukemia in 2006: a perspective. haematologica/the hematology journal | 2006; 91(2): 152-158 [ Links ]
18. Fourouclas N, Campbell PJ, Bench AJ, et al. Size matters: the prognostic implications of large and small deletions of the derivative 9 chromosome in chronic myeloid leukemia. Haematologica 2006; 91: 952-55. [ Links ]
19. Frazer R, Irvine AE, McMullin MF. Chronic Myeloid Leukaemia in The 21st Century. Ulster Med J 2007; 76 (1) 8-17 [ Links ]
20. Fausel C. Targeted Chronic Myeloid Leukemia Therapy: Seeking a Cure. Journal of Managed Care Pharmacy 2007; Vol. 13: 28-212 [ Links ]
21. Isolabella M. Efectos de drogas citotóxicas sobre células HEp-2 y U-937. Universidad de Belgrano 2005; tesis 186 [ Links ]
22. Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. BLOOD 2006;108 (6): 1809-1820 [ Links ]
23. Rozman C, Carreras E. Leucemia mieloide crónica (LMC): un dilema terapéutico. Med Clin (Barc) 1995; 105: 24-26 [ Links ]
24. Calvelo F. Mecanismos de Acción Antitumoral. Boletín Oncológico: http://www.boloncol.com/index2.php?option=com_content&do_pdf=1&id=26.html [ Links ]
25. Or R, Shapira MY, Resnick I, et al. Nonmyeloablative allogeneic stem cell transplantation for the treatment of chronic myeloid leukemia in fi rst chronic phase. Blood 2003; 101: 441-45. [ Links ]
26. Weisser M, Schleuning M, Ledderose G, et al. Reduced-intensity conditioning using TBI (8 Gy), fludarabine, cyclophosphamide and ATG in elderly CML patients provides excellent results especially when performed in the early course of the disease. Bone Marrow Transplant 2004; 34: 1083-88. [ Links ]
27. Crawley C, Szydlo R, Lalancette M, et al. Outcomes of reduced-intensity transplantation for chronic myeloid leukemia: an analysis of prognostic factors from the Chronic Leukemia Working Party of the EBMT. Blood 2005; 106: 2969-76. [ Links ]
28. O'Brien SG. Autografting for chronic myeloid leukaemia. Baillieres Clin Haematol 1997;10:369-88. [ Links ]
29. Nicolau, Magalhães Pessoa L, Sturaro D, Saboya R, Dulley F. Evaluation of early hospital discharge after allogeneic bone marrow transplantation for chronic myeloid leucemia. Sao Paulo Med J. 2007;125(3):174-9. [ Links ]
30. Jabbour E, Cortes J, Giles FJ, O'Brien S, Kantarjian HM. Current and Emerging Treatment Options in Chronic Myeloid Leukemia. American Cancer Society 2007; 2171-2181 [ Links ]
31. Lardon F, Snoeck HW, Berneman ZN, et al. Generation of dendritic cells from bone marrow progenitors using GM-CSF, TNF-alpha, and additional cytokines: antagonistic effects of IL-4 and IFN-gamma and selective involvement of TNF-alpha receptor-1. Immunology 1997; 91:553-559. [ Links ]
32. Choudhury A, Gajewski JL, Liang JC, et al. Use of leukemic dendritic cells for the generation of antileukemic cellular cytotoxicity against Filadelfia chromosome-positive chronic myelogenous leukemia. Blood 1997; 89:1133-42. [ Links ]
33. Chronic Myeloid Leukemia Trialists' Collaborative Group. Interferon alfa versus chemotherapy for chronic mieloide leukemia: a meta-analysis of seven randomized trials. J Natl Cancer Inst. 1997; 89:1616-1620. [ Links ]
34. Kantarjian HM, O'Brien S, Smith TL, et al. Treatment of Filadelfia chromosome-positive early chronic phase chronic myelogenous leukemia with daily doses of interferon alpha and low-dose cytarabine. J ClinOncol 1999;17:284-292. [ Links ]
35. Silver RT, Woolf SH, Hehlmann R, Appelbaum FR, Anderson J, Bennett C, et al. An Evidence-Based Analysis of the Effect of Busulfan, Hydroxyurea, Interferon, and Allogeneic Bone Marrow Transplantation in Treating the Chronic Phase of Chronic Myeloid Leukemia: Developed for the American Society of Hematology. Blood 1999; 94: 1517-1536 [ Links ]
36. McGlave P. Chronic leukemias. In: Beatty PG, ed. Clinical and managed care issues in blood and marrow transplantation for haematological diseases: report of a symposium. March 14, 1996, Washington, DC. Exp Hemat. 1997; 25: 1195-1208. [ Links ]
37. Gale RP, Hehlmann R, Zhang MJ, et al. Survival with bone marrow transplantation versus hydroxyurea or interferon for chronic myelogenous leukemia. Blood 1998; 91:1810-1819. [ Links ]
38. Hansen JA, Gooley TA, Martin PJ, et al. Bone marrow transplants from unrelated donors for patients with chronic myeloid leukemia. N Engl J Med. 1998;338:962-968. [ Links ]
39. Gratwohl A, Hermans J, Goldman JM, et al. Risk assessment for patients with chronic myeloid leukaemia before allogeneic blood or marrow transplantation. Lancet 1998;352:1087-1092. [ Links ]
40. Kujawski LA, Talpaz M. The role of interferon-alpha in the treatment of chronic myeloid leucemia. Cytokine & Growth Factor Reviews 18 (2007) 459-471 [ Links ]
41. Bonifazi F, De Vivo Ao, Rosti G, Guilhot, Guilhot J, Trabacchi E, et al. Chronic myeloid leukemia and interferon-a: a study of complete cytogenetic responders. BLOOD 2001; 98: 3074-3081 [ Links ]
42. Hunter T. Treatment for chronic myelogenous leukemia: the long road to imatinib. The Journal of Clinical Investigation 2007;117: 2036-2043 [ Links ]
43. Braziel RM, Shipp MA, Feldman AL, Espina V, Winters W, Jaffe ES, et al. Molecular diagnostics. Hematology 2003;(1):279-93. [ Links ]
44. Peggs K, Mackinnon S. Imatinib mesylate - the new gold standard for treatment of chronic myeloid leukemia. New Engl J Med 2003;348(11):1048-50. [ Links ]
45. Druker BJ, Moshe T, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. New Engl J Med 2001;344(14):1031-7. [ Links ]
46. Savage DG, Antman KH. Imatinib mesylate - a new oral targeted therapy.New Engl J Med 2002;346(9):683-93. [ Links ]
47. O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003; 348:994-1004. [ Links ]
48. Talpaz M, Goldman J, Sawyers C, Hochhaus A, Silver RT, Douglas Smith BD, et al. High dose imatinib (STI571, Gleevec®) provides durable long-term outcomes for patients (pts) with chronic myeloid leukemia (CML) in accelerated phase (AP) or myeloid blast crisis (BC): follow-up of the Phase II studies [Abstract]. Blood 2003; 102:905a-6a. Abstract no. 3369. [ Links ]
49. Litzow MR. Imatinib resistance: obstacles and opportunities.Arch Pathol Lab Med. 2006; 130:669-679. [ Links ]
50. Kantarjian HM, Talpaz M, Giles F, O'Brien S, Cortes J. New Insights into the Pathophysiology of Chronic Myeloid Leukemia and Imatinib Resistance. American College of Physicians 2006; 913-923 [ Links ]
51. Mauro MJ. Defining and managing imatinib resistance. Hematol Am Soc Hematol Educ Prog 2006:219-25. [ Links ]
52. Corbin AS, La Rosee P, Stoffregen EP, et al. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood 2003; 101(11):4611-4. [ Links ]
53. Apperley JF. Mechanisms of resistance to imatinib in chronic myeloid leukaemia, Part I. Lancet Oncol 2007; 8: 1018-29 [ Links ]
54. Lahaye T, Riehm B, Berger U, et al. Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center: a 4.5-year follow-up. Cancer 2005;103: 1659-1669. [ Links ]
55. Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002;16:2190-2196. [ Links ]
56. Talpaz M, Shah NP, Kantarjian HM, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib resistant Filadelfia chromosome positive leukemias. New Engl J Med 2006; 354(24):2531-2541. [ Links ]
57. Martinelli G, Soverini S, Rosti G, Cilloni D, Baccarani M. New tyrosine kinase inhibitors in chronic myeloid leucemia. Haematologica 2005; 90:534-541. [ Links ]
58. Von Bubnoff N, Peschel C, Duyster J. Resistance of Filadelfia chromosome positive leukemia towards the kinase inhibitor imatinib (STI571, Glivec): a targeted oncoprotein strikes back. Leukemia 2003;17(5):829-38. [ Links ]
59. Hochhaus A, La Rosee P. Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistence. Leukemia 2004;18(8):1321-31. [ Links ]
60. Marcucci G, Perrotti D, Caligiuri MA. Understanding the Molecular Basis of Imatinib Mesylate Therapy in Chronic Myelogenous Leukemia and the Related Mechanisms of Resistance. Clin. Cancer Res 2003; 9: 1248-1252 [ Links ]
61. Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, et al. Detection of the BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood 2003;102(1):276-83. [ Links ]
62. Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Filadelfia-chromosome-positive leukaemias. Lancet Oncol 2003; 4: 75-85 [ Links ]
63. Mohamed AN, Pemberton P, Zonder J, Schiffer CA. The effect of imatinib mesylate on patients with Filadelfia chromosome positive chronic myeloid leukemia with secondary chromosomal aberrations. Clin. Cancer Res., 9: 00-00, 2003. [ Links ]
64. O'Dwyer ME. Chronic myelogenous leukemia. Curr. Opin. Oncol 2003; 15: 10-15. [ Links ]
65. Deininger WN. Optimizing therapy of chronic myeloid leucemia. Experimental Hematology 2007; 35:144-154 [ Links ]
66. Olivieri A. Manzione L. Dasatinib: a new step in molecular target therapy. European Society for Medical Oncology 2007; 18: Supplement 6 [ Links ]
67. Bordessoule D, Turlure P, Trimoreau F, Denizot Y. Sequential mutations causing resistance to both Imatinib Mesylate and Dasatinib in a chronic myeloid leukaemia patient progressing to lymphoid blast crisis. Letters to the Editor / Leukemia Research 2008; 32: 669-677 [ Links ]
68. Martinelli S, Soverini S, Rosti G, Baccarani M. Dual tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia. 2005;19(11):1872-1879. [ Links ]
69. Hampton T. Looking beyond imatinib - next line of targeted drugs for CML shows promise. JAMA 2006;295(4):369-370. [ Links ]
70. Druker BJ, O'Brien SG, Cortes J, Radich J. Chronic myelogenous leukemia. Hematology 2002;(1):111-35. [ Links ]
71. Melo JV, Hughes TP, Apperley JF. Chronic myeloid leukemia. Hematology 2003;17(1):132-52. [ Links ]
72. Cortes J, O'Brien SG, Giles F, Alvarez RH, Talpaz M, Kantarjian H. Investigational strategies in chronic myelegenous leukemia. Hematol Oncol Clin North Am 2004;18(3):619-639. [ Links ]
73. Perkins C, Kim CN, Fang G, Bhalla K. Arsenic induces apoptosis of multidrug-resistant human myeloid leukemia cells that express BCRABL or overexpress MDR, MRP, Bcl-2, or Bcl-xL. Blood 2000; 95(3):14-22. [ Links ]

