










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Universidad Industrial de Santander. Salud
Print version ISSN 0121-0807
Rev. Univ. Ind. Santander. Salud vol.48 no.3 Bucaramanga July/Sept. 2016
https://doi.org/10.18273/revsal.v48n3-2016010
DOI: http://dx.doi.org/10.18273/revsal.v48n3-2016010
Propuesta de formato de reporte de sospecha
de reacciones adversas a medicamentos
por pacientes ambulatorios en Colombia
Daniel Pino Marín1, Pedro Amariles1.
1. Universidad de Antioquia. Medellín, Colombia.
Correspondencia: Pedro Amariles. Dirección: Departamento de Farmacia, Universidad de Antioquia; Calle 70 No. 52-21, Medellín, Colombia.
Correo electrónico: pedro.amariles@udea.edu.co.
Teléfono: +574-2195476
Recibido: 13/11/2015 Aprobado: 29/01/2016 Publicado online: 30/06/2016
Forma de citar: Pino-Marín D, Amariles P. Propuesta de formato de reporte de sospecha de reacciones adversas a medicamentos por pacientes ambulatorios en Colombia. Rev Univ Ind Santander Salud. 2016; 48(3): 365-373.
RESUMEN
Introducción: La obtención de información precisa y oportuna relacionada con las reacciones adversas medicamentosas resulta clave; utiliza estrategia como notificación voluntaria o por un profesional sanitario, ante la autoridad competente. Objetivo: Diseñar una propuesta de formato de reporte de reacciones adversas medicamentosas (RAM) por pacientes ambulatorios en Colombia, basado en recomendaciones de otros países y en requerimientos mínimos definidos para este tipo de formatos. Metodología: Revisión estructurada en PUBMED/MEDLINE de artículos publicados en inglés o español, utilizando los términos Pharmacovigilance AND Patients; y Pharmacovigilance reporting AND patient report. Se incluyeron artículos con información sobre reporte de RAM por pacientes. Con el soporte de la información de la revisión, al igual que con los requerimientos establecidos en las Guías del International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E2A y E2B (R3), se construyó una lista de chequeo, la cual fue confrontada con la revisión de los formatos para el reporte por pacientes de 50 países pertenecientes al Uppsala Monitoring Center. Finalmente, se definieron los ítems a incluir en la propuesta de formato. Resultados: Se identificaron 104 artículos, de los cuales 5 cumplieron con los criterios de inclusión. Se elaboró una lista de chequeo con 34 ítems, la cual fue confrontada con 33 formatos, de los 50 países a los que se pudo acceder y, con ello, se definió el contenido del formato de reporte de RAM por pacientes. Conclusiones: Se diseña y presenta una propuesta de formato de reporte de RAM por pacientes ambulatorios en Colombia, con la información identificada como clave. El formato requiere de un proceso de validación y valoración de su funcionalidad en la contribución al logro de los objetivos de la farmacovigilancia.
Palabras clave: Efectos Colaterales y Reacciones Adversas Relacionados con Medicamentos, Farmacovigilancia, utilización de medicamentos, atención ambulatoria, farmacoepidemiología.
Suggestion of a form for outpatient reporting of
suspected adverse drug reactions in Colombia
ABSTRACT
Introduction: Obtaining accurate and timely information related to adverse drug reactions is key; uses strategy as voluntary reporting by health professionals or to the competent authority. Objective: To design a proposal of format for reporting adverse drug reactions (ADRs) by outpatients in Colombia based on recommendations from other countries and definite minimum requirements for this type of format. Methodology: We completed a structured review in PubMed/MEDLINE database of articles published in English or Spanish, using the terms Pharmacovigilance AND Patients, and Pharmacovigilance reporting and patient report. Articles with information about patients reporting ADR where included. With the support of the information from the review, as with the requirements established in the guidelines from the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) E2A and E2B (R3). We developed a checklist, and then we contrasted the items with the information registered in the format for reporting by patients from 50 countries belonging to the Uppsala Monitoring Center with. Finally, we defined the items to be included in the proposed format. Results: From structured review, we identified 104 articles, of which five met the inclusion criteria. We developed a checklist with 34 items, and then we confronted the 34 items with information registered in 33 of the 50 formats of the countries that we may accessed. We defined the contents of the ADR report format by patients. Conclusions: We design and develop a proposal of ADR reporting format by outpatient in Colombia, which has the information identified as key. The format requires a process of validation and evaluation of its functionality in contributing to achieving the objectives of pharmacovigilance.
Keywords: Drug-Related Side Effects and Adverse Reactions, Pharmacovigilance, Drug utilization, Ambulatory care, Pharmacoepidemiology
INTRODUCCIÓN
La farmacovigilancia es la ciencia y actividades encargadas de la detección, evaluación, entendimiento y prevención de los efectos adversos o cualquier otro problema relacionado con los medicamentos1,2. En este sentido, la obtención de información precisa y oportuna relacionada con las reacciones adversas medicamentosas (RAM) resulta clave. Para ello, como estrategia se utiliza, entre otras, la notificación voluntaria, la cual está basada en la comunicación voluntaria, por parte de un profesional sanitario a la autoridad competente. En dicho reporte se relaciona información que describe una o más RAM en un paciente, al cual se le han administrado uno o varios medicamentos3. Para el reporte, se dispone de un formato de sospecha de RAM que puede variar de país en país, dependiendo de su contexto social y cultural. Sin embargo, el formato debe cumplir los requerimientos mínimos básicos establecidos por el Uppsala Monitoring Center (UMC) y por las Guías del International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E2A y E2B (R3). En el caso de Colombia, se dispone del formato de reporte de sospecha de evento adverso a medicamentos (FOREAM).
La importancia de la notificación espontánea se debe, entre otras, a que: a) es un sistema sencillo y económico, b) no afecta hábitos de prescripción, c) inicia con la comercialización del medicamento y es permanente, d) genera "señales o alertas" a partir de las cuales pueden realizarse estudios más detallados, e) identifica RAM de frecuencia baja (no detectadas en ensayos clínicos), y f) permite obtener datos de todos los ámbitos en los que se emplean los medicamentos (todos los medicamentos, pacientes y médicos)3. Sin embargo, es un sistema que no está exento de limitaciones, siendo el sub-reporte de RAM la más notoria, atribuida, entre otras, a falta de interés, voluntad o disponibilidad de los profesionales encargados de notificar4. Además, la calidad limitada en los reportes, la sub-optimización de los recursos tecnológicos de comunicación y la poca retroalimentación para los profesionales de la salud que realizan los reportes, son obstáculos importantes para la consecución de los objetivos de la farmacovigilancia5,6. En el Reino Unido, se estima un sub-reporte de RAM de medicamentos recién comercializados del 91%7. Por su parte, en Colombia el sub-reporte es cercano al 73%8 y, aunque la prevalencia de RAM en la población ambulatoria no es precisa, se estima en un 6%9.
En el 2010, la legislación europea de farmacovigilancia, con la directiva 2010/84/EU propuso incluir la notificación espontánea por parte de pacientes10,11. Para ello, se estableció la creación de un formato para la notificación por parte de usuarios. De forma similar, la Organización Mundial de la Salud (OMS) en 2011, reconoció que los reportes presentados por usuarios de medicamentos ayudan a la consecución de los objetivos de la farmacovigilancia12. En este marco, una revisión en Pubmed/Medline y EMBASE, orientada a 50 países pertenecientes al UMC, identificó a Colombia como uno de los países suramericanos con un programa de farmacovigilancia avanzado13. Sin embargo, en el país se señala la ausencia de un formato para el reporte de RAM por parte de pacientes14. En este contexto, los objetivos de este trabajo fueron: 1) identificar el contenido mínimo de un formato de reporte de sospecha de RAM por parte de pacientes ambulatorios, y 2) proponer un formato de reporte de RAM por parte de pacientes ambulatorios en Colombia.
METODOLOGÍA
Requisitos mínimos para un formato reporte de sospechas de RAM
Un formato de reporte de sospechas de RAM por parte de pacientes debe cumplir los requerimientos mínimos básicos establecidos por el UMC en las guías ICH-E2A15 e ICH-E2B (R3)16 los cuales son:
- Información que permita identificar al paciente
a) Iniciales, b) genero, c) edad o fecha de nacimiento, d) peso, e) estatura. - Información que permita identificar a quien reporta
a) Nombres, b) dirección, c) número de teléfono, d) profesión. - Descripción de la reacción o evento adverso al uso de medicamentos
a) Descripción completa del evento, b) fecha de inicio de la reacción, c) fecha de finalización de la reacción o duración de esta, d) Información del evento trans y re-exposición del medicamento, e) resultado (outcome). - Información del medicamento sospechoso
a) Nombre comercial, b) nombre genérico, c) número de lote, d) indicación para la cual fue prescrito o usado el medicamento, e) concentración y forma farmacéutica, f) dosis y régimen, g) ruta de administración, h) fecha de inicio del medicamento, i) fecha de finalización del uso del medicamento o duración del tratamiento.
Revisión bibliográfica
Revisión estructurada para identificar propuestas con la información mínima que debe contener un formato para el reporte de sospechas de RAM por parte de pacientes ambulatorios. Para ello, en la base de datos PUBMED/MEDLINE se realizó la búsqueda de artículos publicados en inglés o español, utilizando los términos Mesh: Pharmacovigilance AND Patients, complementada con la utilización de los términos: Pharmacovigilance reporting AND patient report. Se identificaron estudios realizados en humanos y con acceso a texto completo.
Métodos de revisión
Los artículos incluidos fueron seleccionados por 2 investigadores de forma independiente. Para ello, se revisaron los títulos y resúmenes de todos los artículos identificados para decidir su elegibilidad. Los artículos seleccionados se analizaron conjuntamente y, por consenso, se definieron los que fueron incluidos.
Criterios de inclusión
Estudios en los que se encontró información relacionada con el diseño, implementación, revisión de formatos para reporte de sospecha de reacciones adversas por pacientes.
Lista de chequeo
Con base en las guías ICH-E2A15, ICH-E2B (R3)16 al igual que con los resultados de la revisión bibliográfica, se construyó una lista de chequeo en formato Excel, que reunió los elementos necesarios que debe contemplar un formato para el reporte de sospechas de RAM por parte de pacientes ambulatorios.
Revisión formatos internacionales
Se seleccionaron 50 países miembros del UMC, tal como se detalla:
Todos los países de la Unión Europea en los cuales se ha implementado la legislación de farmacovigilancia, más Noruega, Suiza, Rusia, Canadá, E.E.U.U, Argentina, Brasil, Cuba, Méjico, Perú, Israel, Egipto, Irán, China, India, Japón, Oceanía, Australia y Nueva Zelanda, Argelia, Kenia, Marruecos, Nigeria y Suráfrica.
Los formatos para el reporte de sospechas de RAM, por parte de pacientes, fueron obtenidos en la página web oficial del ente regulador de cada país. Los formatos descargados fueron confrontados con la lista de chequeo, por dos investigadores de forma independiente. Los aspectos identificados y no incluidos en la lista de chequeo se describieron en la casilla observaciones.
Construcción propuesta de formato
Los elementos de la lista de chequeo, que en su mayoría fueron contemplados en los formatos internacionales, fueron incluidos en la propuesta inicial del formato para la notificación de RAM por parte de pacientes. Los elementos adicionales fueron definidos por consenso entre los investigadores. Por su parte, con la misma metodología, se identificó y evaluó la pertinencia de la información del contexto de salud colombiano.
RESULTADOS
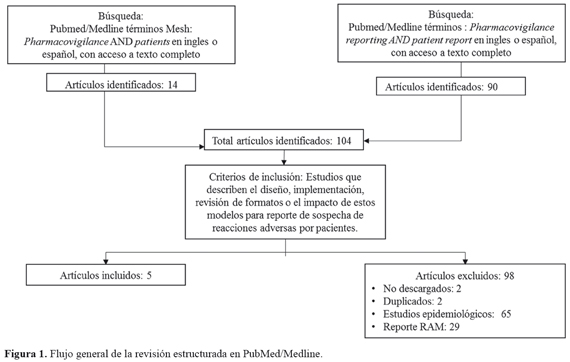
La estrategia de búsqueda "Pharmacovigilance AND Patients" identificó 14 artículos, de ellos uno no tenía disponible el texto completo y, por tanto, se accedió a texto completo de 13. De ellos, 1 aportaba información relevante sobre el contenido mínimo de un formato de reporte de RAM por pacientes. Mientras que, la estrategia "pharmacovigilance reporting AND patient report", identificó 90 artículos, de ellos dos no tenían disponible el texto completo y, por tanto, se accedió a texto completo de 88. De ellos cuatro artículos aportaban al estudio. Por tanto, se identificaron cinco publicaciones que cumplieron los criterios de inclusión. El flujo de la revisión se ilustra en la FIGURA 1.
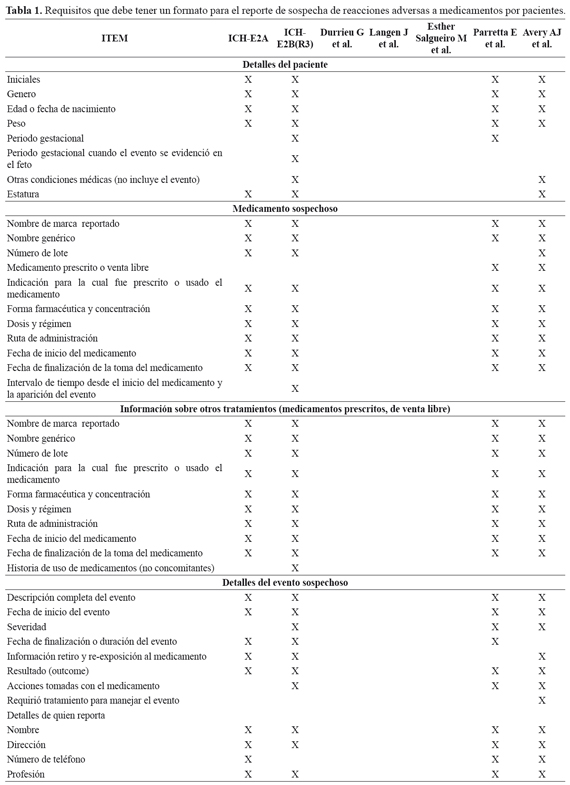
En la TABLA 1 se describen los artículos y su aporte, en la definición de requerimientos mínimos de un formato de reporte de RAM por parte de pacientes ambulatorios. En esta dirección se destacan: a) ICH-E2B (R3)16 considera la presencia del evento en madre/feto, estableciendo la necesidad de información relevante sobre el padre, así no sea el quien presentó el evento. b) Durrieu G, et al.17 refiere que el formato de reporte debe tener la capacidad de establecer una causalidad entre la RAM y el medicamento sospechoso. c) de Langen J, et al.18 establecen que el formato para profesionales sanitarios y pacientes es en esencia el mismo, salvo en el lenguaje usado, debido a que, en el caso de los pacientes, se requiere que el mismo sea de fácil interpretación y comprensión. d) Esther Salgueiro M, et al.19 al igual que de Langen J, et al.18 refieren la similitud entre los formatos de profesionales sanitarios y pacientes. e) Parretta E et al.20 contemplan la posibilidad que el evento se deba a problemas relacionados con el uso de medicamentos; además, consideran el efecto del evento en la calidad de vida del paciente. f) Avery AJ et al.21 buscan establecer relación entre los medicamentos concomitantes y el evento adverso, incluyendo la pregunta: ¿cree usted que este medicamento también podría haber causado el evento?.
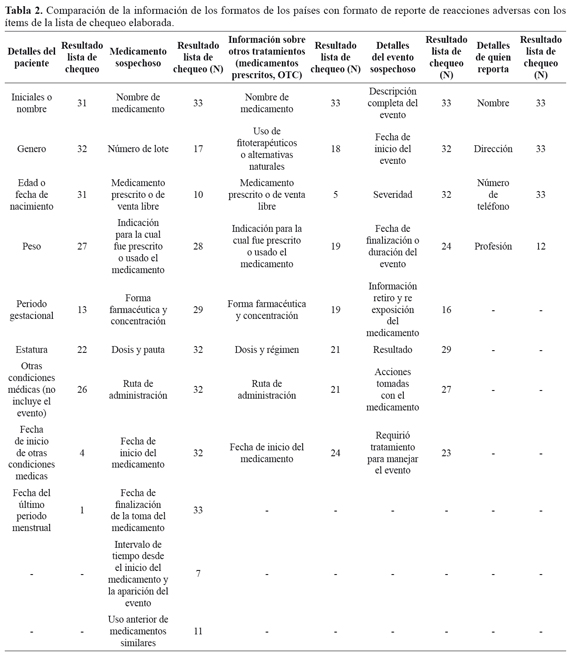
Relacionado con la revisión de los formatos de reporte de sospecha de RAM de 50 países pertenecientes al UMC, se encontró que 36 países cuentan con un formato específico para pacientes. Sin embargo, Cuba no posee el formato en medio electrónico por lo cual no pudo ser analizado; además, Alemania y Noruega requieren la identificación como ciudadano para realizar la notificación. Por tanto, los resultados se presentan con la información confrontada con la lista de chequeo con los 33 formatos obtenidos (TABLA 2). En los formatos de cinco países se incluye la opción de que el paciente informe si considera que el evento está relacionado con un problema de proceso.
DISCUSIÓN
El presente trabajo presenta la información necesaria para la construcción de una propuesta de formato para el reporte de sospecha de RAM por pacientes ambulatorios en Colombia, el cual considera, además de la información y propuestas de otros países, el contexto del sistema de salud de este país.
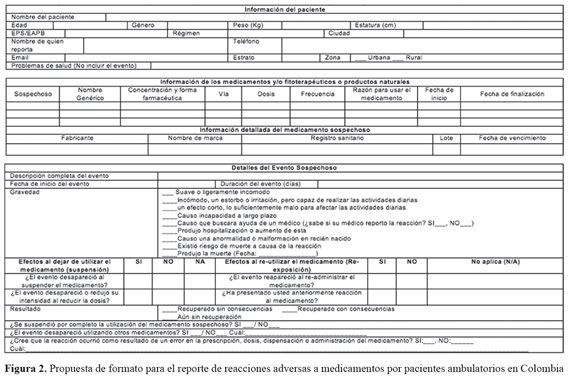
La guía ICH-E2A establece las definiciones y requisitos mínimos de información que debe cumplir un formato de reporte de RAM, orientado al reporte por parte de profesionales sanitarios. Por su parte, la guía para el manejo de la información ICH-E2B (R3)16, es más amplia en su enfoque y considera al paciente como fuente primaria del reporte de sospecha de RAM. Sin embargo, dicha guía no detalla el contenido del formato para el reporte por parte de pacientes. Esta limitación ha sido identificada en algunos estudios18,19. En general, los autores de estos trabajos establecen que, la información para el formato de reporte de sospechas de RAM, debe ser similar en información al formato usado por profesionales sanitarios, buscando una mayor compresibilidad y amigabilidad para los pacientes. En esta dirección, en propuestas como la del Reino Unido21, se identifican algunas diferencias con la propuesta generada en el presente trabajo (FIGURA 2). La principal diferencia radica en que el formato usado en el Reino Unido indaga, de manera opcional, por información sobre el médico tratante. En este sentido, además del nombre y dirección, se pregunta al paciente si autoriza contactar al médico, para obtener información adicional, como resultados de paraclínicos, historia clínica, etc. Además, se pregunta al paciente si el medicamento sospechoso y la medicación concomitante fueron obtenidos por medio de una prescripción médica, en una farmacia o en algún otro lugar. Por su parte, la propuesta de formato para el reporte de RAM por pacientes en Colombia, contempla información propia del contexto de salud y social colombiano, como el nombre de la entidad promotora de Salud (EPS), la afiliación al sistema general de seguridad social en salud (SGSSS), el estrato social y la zona de residencia. Es importante resaltar que, los formatos internacionales y el que se presenta en esta propuesta, están estructurados con preguntas orientadas a obtener el mayor grado de precisión en la información suministrada por el paciente y, por consiguiente, incluyen información exhaustiva y relevante para el análisis de la RAM.24 Por tanto, el resultado son formatos que pueden requerir de un tiempo considerable y, por tanto, pueden resultar desmotivadores para su diligenciamiento.
En general, la notificación por pacientes podría aumentar problemas de reporte selectivo, debido a la influencia de los medios y la publicidad de medicamentos de uso común. Además, podría facilitar el reporte de información trivial o de RAM bien conocidas y definidas, comprometiendo la habilidad del sistema de identificar problemas de seguridad importantes22. Sin embargo, un estudio publicado en Holanda señala que, la diferencia en la información encontrada entre los reportes de pacientes y profesionales sanitarios, radica en que los primeros, están más centrados en el efecto personal de los eventos, mientras que los profesionales sanitarios proveen información clínica detallada. Por ello, estos reportes se complementan y permiten disponer de una mayor información de las RAM25. Además, algunos estudios indican que esta estrategia facilita la identificación de RAM nuevas y que la calidad de los reportes es buena22-23 y, por consiguiente, ayudan a subsanar el sub-reporte de notificaciones. En este sentido, la información proporcionada por los pacientes podría favorecer el reporte de eventos relevantes que los profesionales sanitarios podrían considerar no relacionados con un medicamento, o valorados como poco relevantes, como por ejemplo la disminución en el libido25,26. Por ello, el diseño de un formato de reporte de sospechas de RAM por parte de pacientes ambulatorios en Colombia es una estrategia que podría contribuir al logro de los objetivos de la farmacovigilancia.
El proceso de implementación y ajuste de la propuesta de formato de reporte de RAM por pacientes requiere de la participación de asociaciones de pacientes, buscando identificar e incluir el lenguaje y términos más adecuados; además se debe realizar un estudio destinado a establecer su validación y, con ello, las condiciones deseadas de veracidad, precisión y fiabilidad de la información.
El presente trabajo presenta algunas limitaciones y, por tanto, sus resultados y conclusiones deberían interpretarse con precaución. Entre ellas, cabe destacar que la búsqueda se restringió a la base de datos PubMed/Medline, lo que podría generar un sesgo de información. Además, el resultado de este trabajo es una propuesta preliminar de formato de reporte de RAM por pacientes y, por tanto, el mismo podría resultar demasiado extenso y demandar un tiempo notorio para su diligenciamiento. Por ello, el formato requiere un proceso de validación y adecuación con grupos de pacientes con el fin de establecer el lenguaje adecuado al igual que la posibilidad de depurar información para hacerlo más afable y comprensible.
CONCLUSIONES
Se diseña y presenta una propuesta de formato de reporte de RAM por pacientes ambulatorios en Colombia, con la información identificada como clave. El formato requiere de un proceso de validación y valoración de su funcionalidad en la contribución al logro de los objetivos de la farmacovigilancia.
AGRADECIMIENTOS
Fuente de apoyo financiero: El grupo Promoción y Prevención Farmacéutica recibió financiación de la convocatoria de sostenibilidad 2014-2015 del Comité para el Desarrollo de la Investigación de la Universidad de Antioquia.
CONFLICTO DE INTERÉS
Los autores manifiestan que no existe ningún conflicto de intereses.
REFERENCIAS
1. The importance of pharmacovigilance. World Health Organization & WHO Collaborating Centre for International Drug Monitoring. Geneve: 2002. [ Links ]
2. WHO guidelines on safety monitoring of herbal medicines in pharmacovigilance systems. World Health Organization. Geneve: 2004. [ Links ]
3. Amariles P, Hincapié JA, Jiménez CM, Gutiérrez FJ, Giraldo NA. Farmacoseguridad: farmacovigilancia y seguimiento Farmacoterapéutico. Medellín. 2011. DOI: 10.13140/2.1.2702.8486 [ Links ]
4. Basch E. Systematic collection of Patient-Reported adverse drug reactions: a path to patient-centred pharmacovigilance. Drug Saf. 2013; 36(4): 277- 278. DOI: 10.1007/s40264-013-0026-x. [ Links ]
5. Edwards R. An agenda for UK clinical pharmacology Pharmacovigilance. Br J Clin Pharmacol. 2012; 73(6): 979-982. DOI: 10.1111/j.1365- 2125.2012.04249.x. [ Links ]
6. Organización Panamericana de la Salud. "Buenas Prácticas de Farmacovigilancia". Washington, D. C.: OPS, © 2011. [ Links ]
7. Martin R, Kapoor K, Wilton L, Mann R. Underreporting of suspected adverse drug reactions to newly marketed ("black triangle") drugs in general practice: observational study. BMJ. 1998; 317(7151): 119-120. [ Links ]
8. Calderón C, Orozco J. Ocurrencia de efectos adversos a medicamentos entre el 20 de septiembre y el 2 de octubre de 2004 en el Centro de Atención Ambulatoria Central de la ESE Luis Carlos Sarmiento de Bogotá, Colombia. MED.UIS 2009; 22(2): 127-136. [ Links ]
9. Bustamante C, Calderón C, Castillo I, Cifuentes P, Ramírez J. Reacciones adversas a medicamentos como motivo de ingreso al servicio de urgencias de la Clínica Universitaria Teletón. Cuad Med Invest Salud. 2007; 1: 33-41. [ Links ]
10. THE EUROPEAN PARLIAMENT. DIRECTIVE 2010/84/EU OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 15 December 2010 amending, as regards pharmacovigilance, Directive 2001/83/EC on the Community code relating to medicinal products for human use; 2010. [ Links ]
11. THE EUROPEAN PARLIAMENT. REGULATION (EU) No 1235/2010 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 15 December 2010 amending, as regards pharmacovigilance of medicinal products for human use, Regulation (EC) No 726/2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency, and Regulation (EC) No 1394/2007 on advanced therapy medicinal products; 2010. [ Links ]
12. Vilhelmsson A, Svensson T, Meeuwisse A, Carlsten A. What can we learn from consumer reports on psychiatric adverse drug reactions with antidepressant medication? Experiences from reports to a consumer association. BMC Clin Pharmacol. 2011; 11: 16. DOI: 10.1186/1472-6904-11-16. [ Links ]
13. Margraff F, Bertram D. Adverse Drug Reaction Reporting by Patients: An overview of fifty countries. Drug Saf. 2014; 37(6): 409-419. DOI: 10.1007/s40264-014-0162-y. [ Links ]
14. Hoffmann E, Fouretier A, Vergne C, Bertram D. Pharmacovigilance regulatory requirements in Latin America. Pharm Med. 2012; 26(3): 153-164. DOI: 10.1007/BF03262389. [ Links ]
15. ICH Harmonized Tripartite Guideline: Clinical Safety Data Management: Definitions and Standards for Expedited Reporting E2A. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. October 1994. [ Links ]
16. ICH E2B (R3) Electronic Transmission of Individual Case Safety Reports Implementation Guide - Data Elements and Message Specification; Appendix on backwards and forwards compatibility; availability. February 2014. [ Links ]
17. Durrieu G, Palmaro A, Pourcel L, Caillet C, Faucher A, Jacquet A, et al. French Network of Pharmacovigilance Centres. First French experience of ADR reporting by patients after a mass immunization campaign with Influenza A (H1N1) pandemic vaccines: a comparison of reports submitted by patients and healthcare professionals. Drug Saf. 2012; 35(10): 845-854. DOI: 10.2165/11631800-000000000-00000. [ Links ]
18. de Langen J, van Hunsel F, Passier A, de Jong-van den Berg L, van Grootheest K. Adverse drug reaction reporting by patients in the Netherlands: three years of experience. Drug Saf. 2008; 31(6): 515-524. [ Links ]
19. Esther Salgueiro M, Jimeno FJ, Aguirre C, García M, Ordóñez L, Manso G. Direct reporting by patients of adverse drug reactions in Spain. Farm Hosp. 2013; 37(1): 65-71. DOI: 10.7399/FH.2013.37.1.121. [ Links ]
20. Parretta E, Rafaniello C, Magro L, Coggiola Pittoni A, Sportiello L, Ferrajolo C, et al. Improvement of patient adverse drug reaction reporting through a community pharmacist-based intervention in the Campania region of Italy. Expert Opin Drug Saf. 2014; 13 (Suppl 1): S21-29. DOI: 10.1517/14740338.2014.939582. [ Links ]
21. Avery AJ, Anderson C, Bond CM, Fortnum H, Gifford A, Hannaford PC, et al. Evaluation of patient reporting of adverse drug reactions to the UK 'Yellow Card Scheme': literature review, descriptive and qualitative analyses, and questionnaire surveys. Health Technol Assess. 2011; 15(20): 1-234 DOI: 10.3310/hta15200. [ Links ]
22. Hazell L, Cornelius V, Hannaford P, Shakir S, Avery AJ; Yellow Card Study Collaboration. How do patients contribute to signal detection?: A retrospective analysis of spontaneous reporting of adverse drug reactions in the UK's Yellow Card Scheme. Drug Saf. 2013; 36(3): 199-206. DOI: 10.1007/s40264-013-0021-2. [ Links ]
23. van Hunsel F, Härmark L, Pal S, Olsson S, van Grootheest K. Experiences with adverse drug reaction reporting by patients: an 11-country survey. Drug Saf 2012; 35(1): 45-60. DOI: 10.2165/11594320-000000000-00000. [ Links ]
24. Rolfes L, van Hunsel F, Wilkes S, van Grootheest K, van Puijenbroek E. Adverse drug reaction reports of patients and healthcare professionals-differences in reported information. Pharmacoepidemiol Drug Saf. 2015; 24(2): 152-158. DOI: 10.1002/pds.3687. [ Links ]
25. Härmark L, van Hunsel F, Grundmark B. ADR Reporting by the General Public: lessons learnt from the Dutch and Swedish Systems. Drug Saf. 2015; 38(4): 337-47. DOI: 10.1007/s40264-015-0264-1. [ Links ]
26. Rolfes L, Wilkes S, van Hunsel F, van Puijenbroek E, van Grootheest K. Important information regarding reporting of adverse drug reactions: a qualitative study. Int J Pharm Pract. 2014; 22(3): 231-233. DOI: 10.1111/ijpp.12056. [ Links ]
