











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El Herpesvirus humano de tipo 4 reconocido también como virus de Epstein-Barr, se encuentra clasificado dentro de la familia Herpesviridae, subfamilia Gammaherpessvirinae y género Lymphocryptovirus1 .
El virión maduro presenta un diámetro que va de 120 a 180 nm y su genoma consiste en una doble cadena de ADN linear, dsDNA. Dicho material genético se encuentra envuelto en un core conformado por la nucleocápside icosaédrica de 162 capsómeros la cual es adyacente a una porción de tegumento proteico que la separa de la envoltura viral, esta última caracterizada por presentar espículas glicoproteicas importantes para el reconocimiento de receptores y la posterior entrada a la célula hospedadora2,3. (Figura 1).
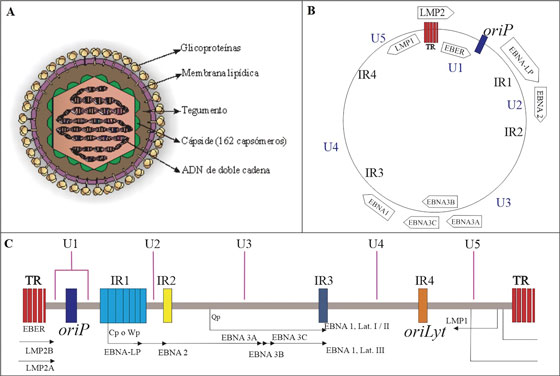
Figura 1 Mofología y estructura genómica del Virus de Epstein-Barr. Adaptado de OK CY, et al78. A) Se muestra la morfología del virus de Epstein-barr. La envoltura contiene varias glicoproteínas que son esenciales para la selección del huésped, el reconocimiento del receptor y la entrada viral a la célula2. B) Forma episomal del genoma viral. C) El genoma del EBV está organizado 85 ORFs, inicialmente basados en la posición y orientación relativa dentro del mapa de enlace del fragmento de restricción BamHI del genoma del ailamiento B95-8, p y posteriormente ampliados tras el secuenciamiento de otros aislamientos virales. Se muestran las secuencias repetitivas terminales (TR) que flanquean el genoma, este se encuentra dividido en cinco dominios de secuencia (U1-U5) por los cuatro dominios de repetición directos internos principales (IR1-IR4). También se muestran las posiciones de la infección latente (oriP) y lıtica (oriLyt) de la replicación del ADN3, 76.
Su material genético contiene aproximadamente 192 kpb que codifican alrededor de 100 genes, organizados en 85 marcos abiertos de lectura u ORFs. El ADN de EBV tiene alto porcentaje G-C y contiene una serie de secuencias repetitivas directas terminales (TR) e intermedias (IR) entre las que se encuentra IRI1 (Secuencia de repetición interna mayor), la cual divide al genoma en una secuencia única larga UL y una corta US. Además, contiene dos orígenes de replicación OriP y OriLyt que son usados en ciclo de latencia y lítico respectivamente4,5,6. Actualmente se conocen dos cepas predominantes de EBV, la tipo 1 o A y la tipo 2 o B ver Figura 1.
Al igual que otros virus de la misma familia, EBV se caracteriza por causar infección latente en el hospedero una vez es adquirido. Aunque en la mayoría de casos la infección cursa de manera asintomática, se ha relacionado con la aparición y desarrollo de diferentes patologías tales como la mononucleosis infecciosa, linfoma de Burkitt, cáncer nasofaríngeo, cáncer gástrico, entre otras.
Replicación viral y patogenia
EBV es transmitido principalmente a través de la saliva donde posteriormente ingresa a la orofaringe, una vez allí, es capaz de infectar tanto a células epiteliales como a linfocitos B vírgenes4 . Al igual que otros herpesvirus, EBV utiliza diferentes combinaciones de glicoproteína y de receptores para definir el tropismo celular, de tal manera que las interacciones necesarias para el ingreso a las células B, varían con respecto al mecanismo de entrada en las células epiteliales como se observa en la Figura 2 7 .
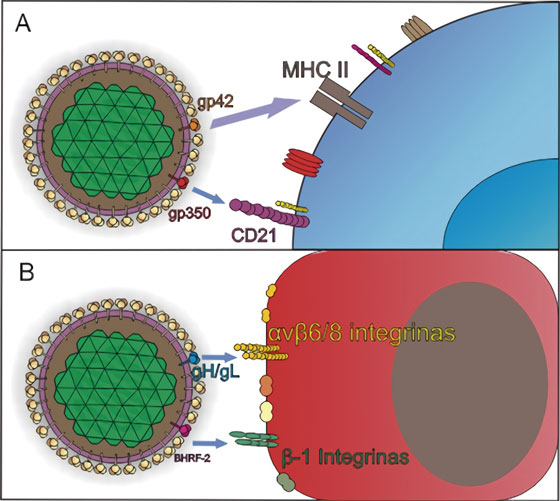
Figura 2 Mecanismos de entrada de EBV a células blanco. Adaptado de Stanfield y Luftig77 . A) La infección de los linfocitos B vírgenes comienza por la unión de la glicoproteína de la envoltura viral, gp350/220 al receptor del complemento tipo 2, o CD21 o al CD35. Tras la fijación del virus a las células B, una segunda molécula, gp42, es escindido en su dominio transmembrana, generando una proteína soluble, que interactúa con su receptor, el Complejo Mayor de Histocompatibilidad clase II, MHC II2. B) En las células epiteliales, la entrada se da por fusión directa con la membrana plasmática del hospedero, y requiere la unión del complejo gH/gL con las integrinas αvβ5, αvβ6 y αvβ8, a través del motivo de unión a integrina KGD, lo que posiblemente induce un cambio conformacional en el complejo, que facilita la unión de gB y desata la fusión2, 77.
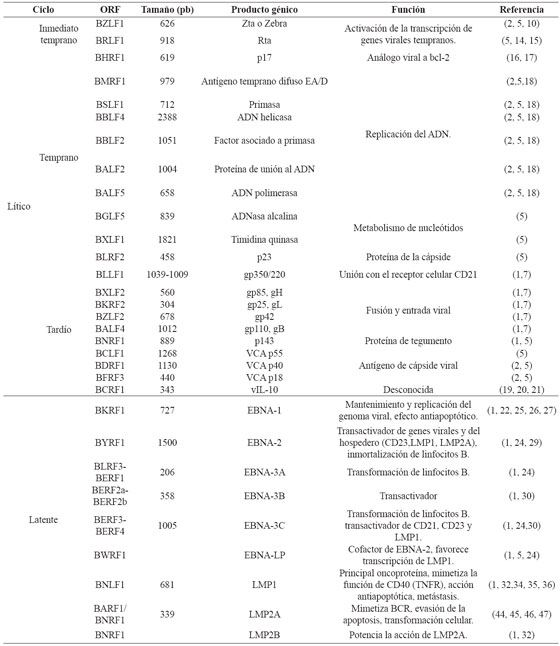
Una vez el virus ha ingresado a la célula, se da lugar el proceso de descapsidación donde el ADN viral es transportado al núcleo y proteínas virales de tegumento son liberadas al citoplasma como BNRF1 la cual se une a la proteína celular Daxx alterando el complejo Daxx/ATRX, facilitando así la transcripción de genes virales. Aunque en ambos tipos celulares el virus puede experimentar un ciclo de replicación lítico, que da como resultado la producción de nuevas partículas infecciosas es en las células epiteliales, a nivel de nasofaringe, donde se ha observado que la replicación toma lugar directamente en la infección primaria. A partir de allí, el virus puede colonizar glándulas salivales, tejidos linfoides y células epiteliales de la orofaringe donde los linfocitos B son infectados a medida que van circulando. Una vez allí, EBV puede llevar una serie de replicación lítica o dar lugar a la expresión diferencial de genes latentes8 . Los productos génicos de las dos fases se exponen en la Tabla 1.
Fase lítica
La fase lítica se presenta durante la primoinfección o la reactivación de la infección y se caracteriza por la expresión de todos los marcos de lectura abiertos del genoma viral5,9. Durante esta fase, el material genético es transportado al núcleo y se replica gracias a la ADN polimerasa viral. Existen tres tipos de productos génicos líticos: inmediatos, tempranos y tardíos5 . Los genes inmediatos BZLF1 y BRLF1, responsables del cambio de fase de latencia a fase lítica, son los primeros en transcribirse. Además, son transactivadores de los genes tempranos y tardíos. BZLF1 codifica para Zta o Zebra, una proteína con tres dominios: un dominio de transactivación, un dominio básico con alta homología a la región conservada de c-jun/c-fos, y un dominio de homodimerización5,10.
Durante la reactivación viral, por medio de Zta y Rta se activa la transcripción desde el promotor de BRLF1 (Rp) a partir de múltiples promotores virales10. Zta también actúa en la replicación al unirse a elementos de respuesta a Zta (Zres), situados dentro del origen de replicación lítico OriLyt. Zres incluyen tanto elementos clásicos de respuesta AP-1, como sitios con motivos CpG metilados que han sido identificados mediante diseños computacionales11,12,13. Por otra parte, Zta contiene un dominio con homología a las ankirinas p53BP2/ASPP2, que se dimeriza formando una estructura denominada ZANK, capaz de interactuar con NF-κβ y p53 inhibiendo las vías de apoptosis mediadas por estas moléculas en linfocitos B, pero estimulándolas en células epiteliales y linfocitos T infectados transitoriamente12 .
BRLF1, codifica para otro transactivador, la proteína Rta, la cual se une directamente a regiones ricas en G-C conocidas como elementos de respuesta a Rta (RREs) ubicados dentro de los promotores de los genes líticos tempranos, donde actúa como potenciador9 , además interactúa con Sp1, MCAF1 y Oct-1, TAF4 y TSG101 para la transcripción de diversos genes virales14 . Rta suprime la inducción del IFN-β, al inhibir la expresión de dos de sus factores reguladores, IRF3 e IRF710 . BZLF1 y BRLF1 también inducen alteraciones morfológicas y funcionales en la mitocondria, obteniendo así el ATP necesario para el ensamblaje y otras funciones relacionadas con la fase lítica15 .
La transcripción de los genes tempranos es inducida por los transactivadores inmediatos, generándose la producción de diversas enzimas, que en su mayoría participan en el proceso de replicación viral5 . Uno de estos genes es BHRF1, el cual codifica una proteína transmembrana de 17 KDa homóloga al prooncogen humano Bcl-2, que actúa como un gen de supervivencia celular al inhibir la apoptosis inducida por estímulos como la γ-radiación, diversos agentes quimioterapéuticos16,17, el retiro del factor de crecimiento, la granzima B y p53, prolongando de esta forma, el tiempo disponible para la replicación de EBV y la producción de nuevos virus16 . Los productos génicos tardíos codifican mayoritariamente proteínas requeridas para el ensamblaje, maduración y liberación del virus18 . Un gen tardío importante para la evasión de la respuesta inmune del hospedador es BCRF1, que codifica un análogo viral de la IL-10 humana, capaz de inhibir la función de las células T, la activación de macrófagos, la síntesis de IFN-γ, y de interferir con la lisis mediada por células NK19,20,21.
En contraste con los genes inmediatos y tempranos, la expresión de los genes tardíos precisa de la replicación viral y la formación de un complejo de preiniciación, en el que BCRF1 se une a la secuencia promotora TATT y a la forma hiperfosforilada de la RNAP II, regulando de esta forma la transcripción de los genes tardíos18,21,
Fase de Latencia
Se caracteriza por tres eventos: el genoma viral persiste de forma episomal principalmente dentro del núcleo de linfocitos B, la replicación está dada por la DNA polimerasa del hospedero, y ocurre una regulación negativa de la expresión génica viral, desencadenando un ciclo de latencias (I - III) asociadas a diversas patologías22 que se muestran en la Tabla 2. Una vez infectado el linfocito B virgen, el virus ingresa en el programa de crecimiento o latencia tipo III, donde expresa la totalidad de sus antígenos induciendo la transformación, multiplicación celular, y activación de linfocitos T citotóxicos. Dependiendo del ambiente externo, características del hospedero y eventos virales, se silencia la expresión de determinados genes, condicionando el paso de un tipo de latencia a otro. Ocasionalmente, las células B de memoria regresan a las amígdalas donde pueden experimentar la diferenciación a células plasmáticas, desencadenando la replicación viral y liberando nuevas partículas a la saliva, para la difusión a otros hospedadores u otras células B como se ilustra en la Figura 3.22,23,24
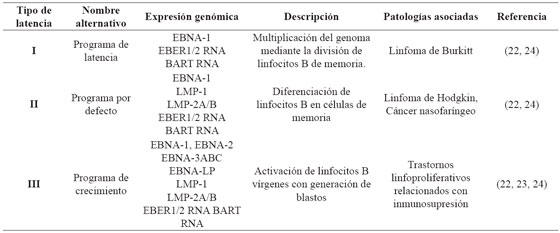
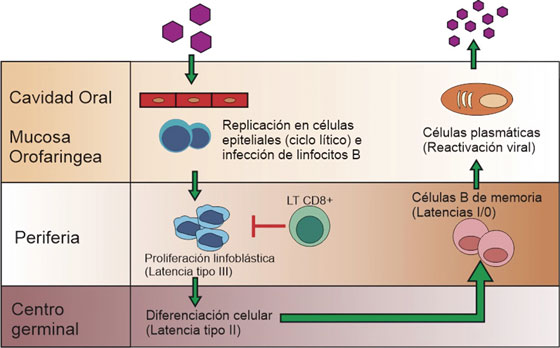
Figura 3 Ciclo del virus de Epstein-Barr. Adaptado de Jha T, et al2 . Una vez infectado el linfocito B virgen, el virus ingresa en el programa de crecimiento o latencia tipo III, donde expresa la totalidad de sus antígenos e induce la transformación y multiplicación celular, generando linfoblastos que al ser altamente inmunogénicos activan la respuesta de los linfocitos T citotóxicos30 . A continuación, tres de las proteínas nucleares virales, regulan negativamente el programa de crecimiento, latencia tipo II, que permite la posterior migración de la célula a los folículos linfoides para iniciar la reacción del centro germinal; una vez generadas las células B de memoria, estas pueden salir a la circulación donde hay un silenciamiento total de la expresión génica, llamada también latencia tipo 0. Eventualmente los linfocitos B de memoria se dividen y así mismo expresan una única proteína nuclear viral, EBNA-1, entrando en latencia tipo I, o migran a las amígdalas reactivando el ciclo lítico2 .
Antígenos Nucleares
El antígeno nuclear 1- EBNA1, es una fosfoproteína de unión específica al ADN, poco inmunogénica, debido a la presencia de triadas de aminoácidos Gly-Gly-Ala, que la protegen de la degradación por proteasoma en la vía dependiente de ubiquitina22,25. Estructuralmente consta de dos regiones ricas en Glicina-Arginina y una en Glicina-Alanina, tiene un sitio de unión a la proteasa de ubiquitina USP7, y un segmento largo C-terminal hidrofóbico de dimerización y unión al ADN. Se transcribe a partir de los promotores Cp y Wp en latencia tipo III, y del promotor ubicado en BamH1Q (Q: Qp) en las latencias tipo I y II. EBNA1 es afín al origen de replicación latente, OriP, cuyos componentes funcionales, el elemento de simetría dioico (DS) y la familia de repeticiones (FR), se encargan de la multiplicación del episoma y la segregación mitótica del ADN viral respectivamente. Cuando se une al elemento FR del OriP, EBNA1 puede transactivar la expresión de otros antígenos nucleares, y de proteínas latentes de membrana, LMPs26 . Además tiene la capacidad de inducir inestabilidad genómica al aumentar los niveles de especies reactivas de oxígeno, y bloquear la interacción de p53 con USP7, disminuyendo y desestabilizando a p5325,27. El silenciamiento de EBNA1 en líneas celulares de Linfoma de Burkitt, se ha visto asociado con la disminución de la proliferación celular, señalando su importancia para la supervivencia de células tumorales.
EBNA5/LP y EBNA2 son las primeras proteínas virales en ser expresadas una vez es infectada la célula, EBNA5, tiene función coactivadora de EBNA-2, cuyo rol en el proceso de transformación celular es conocido. Al no poder unirse directamente al ADN, EBNA-2 secuestra la proteína celular de unión al ADN, RBP-Jκ, para la activación transcripcional de genes celulares como CD23 y virales como LMP1 y LMP2A3,22,26,29. En contraste, la activación de la transcripción que esta mediada por EBNA-2 y EBNA-LP es modulada por los antígenos nucleares 3, EBNA 3A, 3B y 3C, que poseen un origen común y están codificados por fusiones de genes en tándem. Tanto EBNA3A como EBNA3C se asocian a procesos de inmortalización celular, además pueden cooperar con RAS en ensayos de transformación de fibroblastos de roedores, e interrumpir los puntos de control del ciclo celular. EBNA3C, por otra parte, actúa como factor de transcripción induciendo la expresión de CD21, CD23 y LMP126,30,31.
Antígenos de membrana
Proteína latente de membrana 1 (LMP 1): LMP 1 es una proteína integral de membrana de 66 kDa expresada durante los programas de latencia II y III3 . Está conformada por un extremo corto amino-terminal citoplasmático (aminoácidos 1-23) que desempeña un papel en la orientación y procesamiento de la proteína, seis regiones hidrofóbicas transmembrana (aminoácidos 24-186), y una cola citoplasmática carboxi-terminal (aminoácidos 187-386)1,32. LMP 1 se activa constitutivamente a través de mecanismos que involucran la homo-oligomerización y la asociación con balsas lipídicas, y a continuación mimetiza la función del receptor del factor de necrosis tumoral (TNFR), CD40. Los dominios de la región transmembrana (TM) son cruciales para la oligomerización de la molécula y la formación de dominios de señalización en el carboxilo terminal33 , el cual posee la mayor capacidad de señalización, con tres regiones de activación C-terminales (CTAR1, 2 y 3), que proporcionan sitios de acoplamiento para proteínas adaptadoras de señales incluyendo factores asociados a receptor de TNF (TRAFs), proteínas con dominio de muerte asociadas a TNFR (TRADD), el receptor de interacción con proteína quinasa (RIP), BS69 y JAK-334,35.
LMP1 es la principal oncoproteína en la mayoría de tumores EBV positivos. Una de sus características es la capacidad de inhibir la apoptosis en tumores de células epiteliales, así como en linfomas, al estimular la expresión de survivina, TNFAIP3/A20, Mcl-1 y CD137 (en células T y NK), e inhibir por fosforilación a las moléculas proapotóticas Bad y Foxo3a, a través de mecanismos dependientes de las vías de señalización de NF-κβ, AP-1 y PI3K/Akt33,36,37,38. Se ha observado que LMP1 promueve la acumulación nuclear de β-catenina en células de cáncer nasofaríngeo (NPC), aumentando la supervivencia celular a través de la vía Wnt/β-catenina39 . Adicionalmente, posee la capacidad de evadir la apoptosis mediada por metabolitos anticancerígenos como el ácido transretinoico, al inhibir la expresión de su receptor RAR-β2 por metilación del ADN, y disminuir los niveles de caspasas 3 y 9, bloqueando la vía extrínseca de la apoptosis40,41. Por otra parte, LMP1 desempeña un rol importante en la aparición de metástasis al impulsar la sobreexpresión de metaloproteinasa 9, ezrin, TNFAIP2; la activación de ets-1, c-Met y STAT 3, este último por medio de las vías JAK-3 y ERK 1/2 vías que en última instancia conllevan a la pérdida de adhesión celular, degradación de la matriz extracelular y aumento de la motilidad36,37,42,43.
Proteína latente de membrana 2 (LMP2): LMP2 codifica las proteínas LMP2A y LMP2B, transcritas a partir de promotores diferentes en las repeticiones terminales fusionadas del episoma viral. LMP2A y LMP2B poseen 12 segmentos transmembrana y una cola carboxilo-terminal, además LMP2A cuenta con una secuencia adicional amino- terminal citosólica de 119 aminoácidos con capacidad de señalización32 .
La cola amino terminal se encuentra constitutivamente fosforilada dentro de múltiples sitios, incluyendo dos regiones ricas en tirosina (Y74/Y85) que constituyen motivos de inmunoreceptores activables por tirosina (ITAM), donde la proteína Syk es secuestrada y activada; una tirosina en 112 que une y activa la tirosina quinasa Lyn32,44, y dos motivos ricos en prolina (PY), responsables de la unión de Ubiquitina-ligasas U3 y la activación de la β-catenina en células epiteliales al inhibir la GSK-3 por medio de la proteína quinasa C, señalizando e inhibiendo la diferenciación de células epiteliales45 .
LMP2A mimetiza la función del receptor de célula B (BCR), uniendo inicialmente a Lyn, seguida de Syk y desencadenando la activación de la proteína de unión a célula B (BLNK), y las vías NF-κβ, MAPK/ERK y Ras/PI3K/Akt, a través de las cuales promueve la supervivencia de células B, al inducir los genes antiapoptóticos Bcl-2 y Bcl-xL, lo que parece ejercer una función crítica en la infección temprana (4 a 7 días)44,46. Por tanto, al actuar como un BCR activado, LMP2A aumenta la activación temprana, proliferación y supervivencia de células B, favoreciendo el establecimiento de LCLs. LMPA2 también es capaz de interactuar con el residuo 33 de WOX-1 un supresor tumoral, a través de sus regiones PY, desatando la activación de ERK1/2 y cascada abajo el factor de transcripción Fra-1, lo que conduce a un aumento en la expresión de metaloproteinasa (MMP) 9, proteína implicada en la invasión celular47 .
Productos génicos no codificados: EBER 1 y EBER 2 son pequeños ARNs no poliadenilados de 167 y 172 nucleótidos respectivamente, se transcriben a partir de la polimerasa III del hospedero y son altamente expresados en las células en latencia, ambos tipos tienen una estructura secundaria bien definida que comprende el apareamiento intermolecular de bases y la formación de bucles. Los EBERs tienen la capacidad de interactuar con diferentes moléculas del hospedero, tales como el antígeno del lupus (La), el ARN dependiente de proteína quinasa R (PKR), la proteína ribosomal (L22), el ácido retinoico inducible del gen I (RIG-1) y al factor AUF-1; dichas relaciones se asocian con el crecimiento y proliferación celular, se ha reportado que los EBERs confieren resistencia celular a la apoptosis dependiente de PKR, la inducción de citoquinas y la modulación del sistema inmune innato22,48.
Patologías asociadas, diagnóstico y tratamiento
Mononucleosis infecciosa
En países en vía de desarrollo la infección por el virus de Epstein-Barr se da principalmente durante la infancia, cursando de manera asintomática, mientras en países desarrollados, ocurre durante la adolescencia o la adultez, asociada a la aparición de enfermedad en el 50% de los casos, cuatro a seis semanas después del contacto viral49 . La Mononucleosis infecciosa se caracteriza por la proliferación del tejido linfoide y leucocitosis mononuclear con linfocitos atípicos. Presenta un inicio insidioso, seguido de fiebre, faringitis, dolor de garganta y linfoadenopatía cervical posterior, entre otros síntomas.
La primoinfección por el virus de Epstein-Barr desencadena una respuesta inmune que es responsable en gran medida de la sintomatología, inicialmente los virus provocan una potente respuesta de interferones tipo I y la intervención de receptores tipo toll tales como TLR2, TLR3, TLR7 y TLR9. Dentro de las citoquinas proinflamatorias detectadas en suero de pacientes con mononucleosis infecciosa destaca el INF-γ, IL-2, IL-6, IL-1β y TNF-α. La respuesta adaptativa juega un rol fundamental tanto en el control de la infección como en la presentación de los síntomas, los linfocitos atípicos o células de Downey, son linfocitos T citotóxicos activados que responden a la infección por EBV, caracterizados por poseer un mayor tamaño que los linfocitos maduros y por tener un citoplasma vacuolado basófilo, aunque su hallazgo no es patognomónico50,51,52,55.
El diagnóstico de la infección aguda por EBV se basa en la presentación clínica y en los hallazgos de laboratorio, las pruebas serológicas demuestran la presencia de anticuerpos heterófilos en el 60 – 80% de los casos mediante el ensayo de Paul-Bunnell, el virus induce también el desarrollo de anticuerpos específicos, de importante determinación cuando el paciente manifiesta presentaciones clínicas atípicas o anticuerpos heterófilos negativos. La IgM dirigida contra los antígenos de la cápside viral (VCA anti-IgM), se detecta en el 90% de los pacientes hacia el séptimo día de la aparición de los síntomas, mientras la IgG anti VCA tiene un pico entre el segundo y cuarto mes de la infección y persiste de por vida. Los anticuerpos dirigidos contra EBNA-1 se generan hasta tres meses después del contacto viral, siendo del tipo IgG. Dado que en individuos inmunocompetentes el proceso de linfoproliferación es autolimitado, el tratamiento es principalmente sintomático mediante el uso de analgésicos y antipiréticos, sin embrago se han empleado algunos medicamentos antivirales como aciclovir y ganciclovir y también se ha hecho uso de corticoesteroides53,54,55,56.
Linfoma de Burkitt
El linfoma de Burkitt (LB) es un linfoma no Hodgkin de células B, altamente proliferativo, que presenta tres variantes clínicas: endémica, esporádica y asociada a inmunodeficiencia, similares en morfología, inmunofenotipo y características genéticas. La variante endémica ocurre en áfrica ecuatorial y Papua Nueva Guinea, donde representa el 90% de todos los linfomas y en la cual EBV es encontrado en el 96% de los casos56,57.
Histológicamente se caracteriza por la presencia de células monomórficas de tamaño medio que presentan un núcleo aumentado de tamaño, redondo a oval con un patrón de cromatina dispersa y conteniendo múltiples nucléolos basófilos paracentrales, cuyo inmunofenotipo se caracteriza por fuerte expresión de inmunoglobulinas, CD20, CD19, CD22, CD10, Bcl-6, Y Ki-67, y bajos niveles de MHC I, además de nula expresión de CD23, CD5 y Bcl-256,58,59.
Los sitios de localización tumoral están relacionados con la variable clínica, así, en la forma endémica son frecuentes las presentaciones faciales (mandibular, periorbital) y abdominales, mientras que la afectación del ovario y médula ósea es infrecuente. Por otra parte, en la forma esporádica, el 80% de los casos presentan afectación abdominal, seguida de la presentación en cuello y cabeza. Alrededor del 20% de los pacientes en países desarrollados presentan infiltración de la médula ósea clasificada en algunos casos como un síndrome leucémico59,60.
El evento clave en la patogénesis de LB es la adquisición de una translocación cromosomal del protoncogén c-Myc desde el brazo largo del cromosoma 8, a los loci de las cadenas pesada o ligera de las inmunoglobulinas en los cromosomas 2, 14 o 22, generando una expresión constitutiva del gen. EBV contribuye a la generación de LB bien sea aumentando la cantidad de accidentes genéticos, dando lugar a la translocación, o complementando la actividad de c-Myc61 . Las células tumorales infectadas con EBV muestran una latencia tipo I, en la cual EBNA 1 coopera con c-Myc desrregulado principalmente a través de tres de sus funciones: antagonizando a p53, evadiendo el reconocimiento por los linfocitos TCD8+ y manteniendo el episoma viral61,62.
En células de LB, se ha encontrado que tanto la presentación de antígeno por MHC I, como la respuesta citotóxica son ineficientes. EBNA 1 participa en la evasión de la eliminación por linfocitos T CD8+, mediante la formación de un complejo con el proteasoma, bloqueando la degradación proteica y por lo tanto, frustrando el procesamiento y presentación en moléculas MHC61,62,63.
La prueba diagnóstica más común es el exámen citológico de un aspirado con aguja fina, acompañado de pruebas complementarias como radiografía de torax59,64,65. Un algoritmo alternativo propone el uso de un panel de seis anticuerpos – CD20, CD10, BCL-2, Ki-67, CD38 y CD44, distribuidos en tres fases. Las fases 1 y 2 utilizarían inmunotinciones, que detectarían el 90% de casos y la fase 3 utiliza FISH, detectando el porcentaje restante66 .
El tratamiento estándar contra el linfoma de Burkitt consiste en regímenes específicos que incorporan cursos intensivos de quimioterapia con agentes alquilantes fraccionados y agentes específicos de fase capaces de cruzar la barrera hematoencefálica. El tratamiento primario en niños consiste en la administración de COPADM (ciclofosfamida, vincristina, prednisolona, doxorrubicina, metotrexato) con dosis aumentadas de metotrexato y adicion de citarabina en los pacientes con estadíos superiores al B, sin embargo, este régimen tiene un alto grado de toxicidad, por lo cual diferentes protocolos han experimentado con dosis reducidas67,68,69. En adultos, el tratamiento incluye el uso de rituximab. Se encontró que el uso combinado de ciclofosfamida, etopósido, doxorrubicina, vincristina, prednisona y rituximab (EPOCH-R) en dosis disminuidas fue altamente efectivo para el tratamiento de adultos con LB esporádico o asociado a VIH, lográndose una menor incidencia de neutropenia y otros efectos tóxicos que con la administración de dosis estándar70,71.
Carcinoma Nasofaríngeo
La transformación tumorgénica del epitelio nasofaríngeo se presenta principalmente de los 40 a 60 años, siendo endémica en el sur de China, donde afecta 1-40 de cada 100.000 personas. El compromiso de las condiciones inmunes, sumado a un microambiente inflamatrio crónico, podría contribuir a la patogenia de EBV en el desarrollo de tumores malignos. Histológicamente se puede presentar como carcinoma escamoso queratinizante, tipo I; carcinoma escamoso no queratinizante, tipo II; y carcinoma indiferenciado, tipo III, que corresponde al patrón histológico asociado a EBV. Epistaxis, otitis, secreción y obstrucción nasal pueden ser algunas de las primeras manifestaciones clínicas, otros síntomas incluyen dificultad para respirar, linfoadenopatía cervical y el compromiso de nervios craneales72 .
Es común que la infección en células epiteliales tanto de orofaringe como de nasofaringe desencadene la fase lítica viral, sin embargo EBV también puede establecer latencia, donde LMP1 y otras proteínas expresadas en latencia II promueven la iniciación y progresión tumoral. Estudios realizados in-vitro señalan que la infección por EBV induce la detención del crecimiento en el epitelio de la nasofaringe, sin embargo la sobreexpresión de ciclinas (D1 principalmente) y la disminución de p16 contrarrestan la detención del crecimiento, además la sobreexpresión de múltiples subunidades de los factores de transcripción NF-κB, juega un papel importante en la desregulación necesaria para la progresión tumoral32,73.
Muchos de los pacientes con carcinoma nasofaríngeo se presentan con una fase avanzada de la enfermedad, más de seis meses después del inicio de los síntomas, resultando en un mal pronóstico, las herramientas diagnósticas incluyen exámenes de cabeza y cuello junto con la evaluación de la función del nervio craneal. La nasoendoscopia es imprescindible para el diagnóstico, el monitoreo de la función auditiva puede contribuir a direccionar el diagnóstico dado que se ve afectada por la presión que ejerce el tumor. Una de las herramientas más ampliamente utilizadas es el examen radiológico y tanto la tomografía computarizada como la resonancia magnética son también empleadas. La radioterapia es el tratamiento primario para carcinoma nasofaríngeo en todas las etapas de la enfermedad74,75.

