











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Las Alteraciones del Desarrollo Sexual (ADS) son consideradas como un conjunto de condiciones que afectan la determinación y/o diferenciación sexual y que pueden diagnosticarse en cualquier etapa de la vida, desde la formación intrauterina hasta la adultez. Su identificación se realiza al momento del nacimiento cuando se evidencia un fenotipo de órganos genitales externos con una configuración alterada1. Las ADS también pueden ser consideradas como el conjunto de condiciones genéticas en donde el desarrollo del sexo cromosómico, gonadal o anatómico es atípico1),(2.
En general, la prevalencia de las ADS con ambigüedad genital es aproximadamente 1 en 5000 nacimientos1),(3. Sin embargo, existen otras malformaciones genitales congénitas mucho más frecuentes y que se clasifican dentro del espectro de ADS como alteraciones menores o inclasificadas; por ejemplo, la criptorquidia considerada como la alteración genital más frecuente en los varones recién nacidos con una incidencia aproximada de 1.6 a 9%4. Las hipospadias tienen una prevalencia difícil de estimar dado que varía según las regiones en el mundo, se estima que puede ir de 2-43. Dos casos por cada 10.000 nacimientos5),(6.
El tema es importante porque puede constituir una urgencia tanto a nivel endocrinológico como social. Un Recién Nacido (RN) puede terminar en una posible crisis adrenal como consecuencia de una Hiperplasia Adrenal Congénita (HSC), con un alto riesgo de morbimortalidad7. Adicionalmente, se pueden cometer errores en la asignación del sexo del RN, que podrían repercutir negativamente a nivel familiar, social y legal. Por otra parte, es conveniente que los profesionales en el área de la salud utilicen correctamente la nomenclatura, y de este modo evitar términos considerados controversiales y en ocasiones, hasta despectivos. Teniendo en cuenta lo anterior, el objetivo de esta revisión es realizar un abordaje integral de las ADS y reconocer la importancia de los equipos transdisciplinarios para el manejo de esta patología.
Metodología
La siguiente revisión incluye una búsqueda bibliográfica de la literatura científica biomédica, llevada a cabo empleando los siguientes términos MeSH: Disorders of Sex Development, Newborn, Ovotesticular Disorders of Sex Development, True Hermaphroditism, Gonadal Dysgenesis, Adrenal Hyperplasia, Congenital. La búsqueda se realizó en cinco bases de datos electrónicas: National Library of Medicine database (PUBMED/MEDLINE), Scientific Electronic Library Online (SciELO), Google Scholar, Literatura Latinoamericana de Información en Ciencias de la Salud (LILACS) y Clinical Key; se limitó la búsqueda para artículos en idioma español o inglés de los últimos 10 años. Se obtuvieron artículos originales, presentación de casos, consensos y artículos de revisión, los cuales 35 fueron incluidos en esta revisión.
Fisiología del desarrollo sexual normal
El desarrollo sexual ocurre durante la vida fetal y se presenta en dos fases, la primera es la “determinación del sexo” en donde se produce la diferenciación de la gónada bipotencial ya sea testículo u ovario. La acción de genes como SF1, WT1, SOX9 y DAX1 son requeridos para la formación de la gónada bipotencial. Posteriormente, ocurre la fase de “diferenciación del sexo”, en donde a través de la acción hormonal de las gónadas se produce el desarrollo de la configuración de los genitales externos, ya sea en masculinos o femeninos3),(8),(9.
Desarrollo sexual masculino
La acción del gen SRY (que reside en el brazo corto del cromosoma Y) conduce al desarrollo de los testículos a las seis semanas de gestación8),(9. Cerca de la semana ocho, comienza la producción de Hormona Antimülleriana (HAM) por las células de Sertoli que conduce a la regresión de los conductos de Müller (útero, parte superior de la vagina y trompas de Falopio). Las células de Leyding son estimuladas por la gonadotropina coriónica humana para producir andrógenos, posteriormente esta función es asumida por la hormona luteinizante, producida en la glándula pituitaria durante el segundo y tercer trimestre. Durante el primer trimestre, la presencia de andrógenos conduce al desarrollo de los conductos de Wolff para formar las estructuras genitales internas masculinas (vasos deferentes, vesículas seminales y epidídimos)3. En la semana 12 de gestación, la virilización en el feto masculino se debe a la producción de la testosterona, la cual es convertida a Dehidrotestosterona (DHT), por la acción de la enzima 5a-reductasa, que posteriormente conducirá a la pigmentación, la rugosidad y la fusión de los pliegues labioescrotales, el crecimiento del pene y la migración de la uretra a la punta del glande. Por último, el descenso testicular ocurre en la última mitad del tercer trimestre de embarazo1),(3),(9.
Desarrollo sexual femenino
Las acciones de los genes en el cromosoma X y genes autosómicos como WNT4, Respondina, Folistatina y FOXL2 están implicados en el desarrollo ovárico1),(3. El gen WNT4 permite la formación de las estructuras mullerianas, la diferenciación del útero y el cérvix en la semana 12 de gestación3. La secreción de estrógenos por el ovario no es necesaria para el desarrollo genital interno o externo de la mujer. La diferenciación genital femenina se produce debido a la ausencia de testículos, en donde la falta de HAM hace que las estructuras müllerianas persistan y la no producción de andrógenos gonadales conduce a la regresión de los conductos de Wolff y permite que se formen los labios menores y mayores y la parte inferior de la vagina1),(3),(9.
Nomenclatura
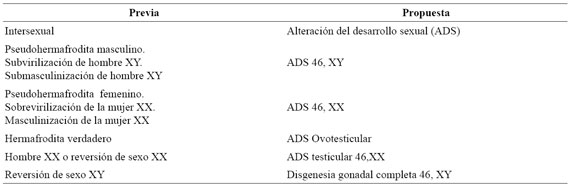
Los avances en el área genética y su correlación con la clínica en pacientes con ADS, han permitido que ciertos términos, algunos considerados confusos y/o estigmatizadores, hayan sido renovados por otros más adecuados2. En el año 2005, se elaboró una Declaración de Consenso para el manejo de las ADS, allí se propuso la utilización de una nueva terminología que permitió abolir términos como “hermafrodita”, “pseudohermafrodita” “intersexo” y “reversión de sexo”. La propuesta utilizaría un algoritmo basado en la constitución cromosómica sexual acompañado de la frase “Disorders of Sexual Development” (DSD de sus siglas en inglés)3),(10),(11. En países europeos se han realizado estudios que comprueban que los pacientes y familiares, han comenzado a emplear la terminología recientemente propuesta con buena aceptación12), sin embargo, no hay estudios para Latinoamérica. En la siguiente tabla se describe el cambio propuesto para la nomenclatura de esta patología:
(Tabla 1) Cambio de nomenclatura de las alteraciones del desarrollo sexual. Declaración de consenso 20052.
Clasificación
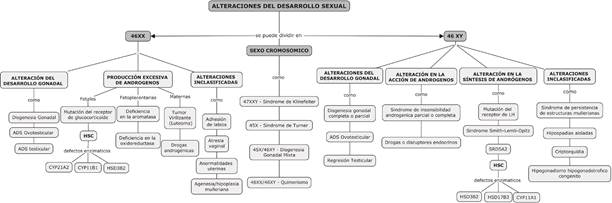
Para realizar la clasificación de las ADS se sigue el esquema aportado por el Consenso de Alteraciones del desarrollo sexual del 201810 (Figura 1).
Fuente: Realizada por autores.
Figura 1 Siglas ADS: Alteraciones del Desarrollo Sexual. HSC: Hiperplasia Suprarrenal Congénita. CYP21A2: Enzima 21 hidroxilasa. CYP11B1: Enzima 11-b hidroxilasa. HSD3B2: Enzima 3-beta hidroxiesteroidedeshidrogenasa 2. HSD17B3: Enzima 17-beta hidroxiesteroidedeshidrogenasa. CYP11A1: Escisión de la cadena lateral de colesterol. SRD5A2: 5 alfareductasa 2
El grupo etiológico más común es ADS 46XX13),(14. En general, la HSC y la Disgenesia Gonadal (DG) constituyen alrededor del 50% de todos los casos de ambigüedad genital neonatal13),(15),(16. A continuación, se describen algunas de estas condiciones según la clasificación propuesta:
Hiperplasia suprarrenal congénita: Es la causa más común en pacientes 46XX7),(14),(15. Es una enfermedad autosómica recesiva en donde hay una deficiencia en la síntesis de esteroides adrenales. La incidencia mundial estimada es de aproximadamente 1:5000-15000 nacidos vivos7),(16. Cerca del 90-95% de las HSC se deben a déficit de la enzima 21 hidroxilasa, que no permite la conversión de 17-hidroxiprogesterona por lo que se acumula este metabolito7),(14),(17. En general, se produce una disminución en la síntesis de glucocorticoides y mineralocorticoides. Por retroalimentación negativa, la hipófisis sigue enviando ACTH para estimular la glándula suprarrenal, en consecuencia, los precursores de esteroides toman la vía de la síntesis de andrógenos, que conlleva a su sobreproducción1),(17. Cuando hay deficiencia de la enzima 21 hidroxilasa, la variabilidad de los procesos de virilización de los genitales externos de pacientes 46XX va a depender del grado de funcionalidad enzimática; sus genitales internos no se verán afectados dado que la condición no se relaciona con la presencia de tejido testicular que pueda producir HAM. En pacientes 46XY, los genitales externos se mantienen con apariencia normal dada la influencia de andrógenos17),(18. Una deficiencia severa de enzimas (<2%) provoca una crisis de pérdida de sal alrededor de la segunda semana de edad debido a la deficiencia de mineralocorticoides1),(17.
ADS ovotesticular: Representa del 3 al 10% de todos los casos de ADS, la incidencia estimada es alrededor de 1 en 100.000 nacimientos15),(16,)(19. La ADS ovotesticular ocurre cuando hay tejido ovárico y testicular dentro de una gónada (ovotestes) o en gónadas opuestas. El cariotipo más frecuente es el 46 XX en un 60%, seguido por los mosaicismos14),(19. Fenotípicamente, las estructuras genitales internas y externas son variables y dependen del tejido gonadal predominante1),(13),(15. El riesgo de malignizaciónes muy bajo (<5%) para pacientes 46XX2),20 y del 10% para pacientes con cariotipo 46XY o mosaicismos19.
Disgenesia gonadal: La incidencia estimada es 1:10.000 nacidos vivos16 y esta condición supone una función inadecuada de la gónada bipotencial. Clínicamente en general, los pacientes 46XY con DG reciben atención temprana por una evidente alteración en el fenotipo; en 46XX, la apariencia de los genitales externos suele estar normal, en ellas puede sospecharse por pubertad retardada1.Esta condición puede dividirse en Disgenesia Gonadal Completa (DGC), Disgenesia Gonadal Parcial (DGP) y Disgenesia Gonadal Mixta (DGM). En DGC, las gónadas son inactivas bilateralmente de forma temprana o tardía; la mayoría de pacientes 46XY con DGC tienen genitales internos y externos femeninos con algún grado de ambigüedad y presentan incapacidad de desarrollar características sexuales secundarias, esto debido a la ausencia de testosterona. La DGP ocurre cuando una o ambas gónadas producen una cantidad deficiente de hormonas; estos pacientes pueden presentarse desde hipospadias hasta ambigüedad genital y el grado de virilización y regresión mulleriana van a depender de la función testicular. La DGM ocurre cuando una gónada es completamente inactiva y la otra tiene DGP o función normal. En pacientes 46XY con DGM, la regresión de la estructura mulleriana es típicamente ipsilateral a la gónada en funcionamiento13),(21.
ADS testicular: Se produce cuando el gen SRY se agrega a un cromosoma X mediante translocación1. Clínicamente se caracteriza por un fenotipo masculino con testículos pequeños, falla testicular primaria, azoospermia y ginecomastia21.
Síndrome de insensibilidad androgénica: Existe una mutación del gen receptor de andrógenos ligada a X, por lo que afecta a los pacientes 46XY1),(21. La incidencia es 1:10.000-20.000 nacimientos22. En el Síndrome de Insensibilidad Completa a los Andrógenos (CAIS), no hay acción de la testosterona ni de la de DHT sobre el receptor, por lo que la virilización de los genitales externos, el desarrollo de los conductos de Wolff y el crecimiento del vello están completamente ausentes, resultando un fenotipo femenino11),(14. Adicionalmente, tienen regresión de las estructuras mullerianas debido a la presencia de tejido testicular que producirá HAM22. Se sospecha por la presencia de gónadas en el canal inguinal, amenorrea primaria o infertilidad. En el Síndrome de Insensibilidad Parcial a los Andrógenos (PAIS), la apariencia de los genitales externos puede ser muy variable, dependiendo del grado de funcionalidad del receptor androgénico22),(23. Pueden presentarse como hipospadias con testículos inguinales o criptorquidia aislada bilateral y micropene(14),(23). El Síndrome de Insensibilidad Leve a los Andrógenos (MAIS) puede presentarse como espermatogénesis deteriorada aislada, alteración de la virilización observada en la pubertad, impotencia y/o casos de ginecomastia14),(22.
Síndrome Turner: Presentan clásicamente un cariotipo 45X, resultado de la no disyunción del cromosoma X durante la meiosis. Sin embargo, también pueden presentar un cariotipo en mosaico con presencia de más líneas celulares1. La incidencia estimada se encuentra en 1:2500 nacimientos16. Clínicamente, desde el nacimiento presentan talla baja (debido a la haploinsuficiencia del gen SHOX), linfedema, cuello alado, paladar alto, cuartos metacarpianos cortos, nevos múltiples pigmentados y displasia ungueal, asociados con anomalías en órganos como corazón y riñones. Pueden tener DGC que impide espontáneamente el inicio de pubertad; otros comienzan el desarrollo puberal, pero no logran alcanzar el desarrollo sexual completo debido a la falla gonadal1),(22.
Síndrome de Klinefelter: El cariotipo clásico es 47XXY, aunque se puede presentar con más adiciones del cromosoma X1. La incidencia es alrededor de 1: 500-1000 hombres16),(22) y es la causa más común de infertilidad masculina. Presentan estatura alta y desproporción de segmentos con brazos y piernas largas, asociado a un retraso en el desarrollo social13 y dificultades en el aprendizaje16. Presentan hipogonadismo hipergonadotrópico debido a un fallo testicular, con gónadas firmes y pequeñas, y micropene1),(22.
Evaluación diagnóstica
Los RN con ambigüedad genital deben evaluarse oportunamente siguiendo un algoritmo, sin embargo, en algunas ocasiones el diagnóstico es en la adolescencia por clitoromegalia, presencia de gónadas en labios mayores, virilización progresiva, infertilidad, retardo puberal, amenorrea primaria o hernias inguinales en mujeres y en varones hematuria cíclica o desarrollo de mamas8),(16),(22),(24.
Anamnesis
En la HSC se pueden presentar síntomas como vómitos a repetición, falla de medro, hipotonía, hipoglucemia, somnolencia/letargia, irritabilidad, convulsiones o incluso presentar una crisis adrenal.
Se sugiere preguntar datos como 1) Historia de consanguinidad entre padres, que orientara hacia la presencia de posibles trastornos recesivos como HSC, CAIS o trastornos cromosómicos. 2) Historia de familiares de primer grado con presencia de ADS como criptorquidia/hipospadias, casos de infertilidad o amenorrea primaria 3) Casos de muertes neonatales no explicadas ante la sospecha de HSC 4) Virilización materna en el embarazo o exposición prenatal a tóxicos, a fármacos o a disruptores endocrinos, por ejemplo, fármacos androgénicos como progesterona o testosterona8),(16),(22.
En los RN con bajo peso al nacerse presenta con más frecuencia las hipospadias; y los casos de criptorquidia están relacionados con RN pretérmino, posiblemente por la falta del descenso gonadal que ocurre en la fase final del último trimestre3),(10),(24.
Examen físico
En el examen de los genitales externos se debe emplear términos intermedios, que no orienten hacia la asignación de un sexo. Es recomendado utilizar términos como tubérculo genital y gónadas, evitando hacer mención palabras como pene, testículos, clítoris, vagina, etc.
Tubérculo genital: evaluar medidas de longitud, presencia de cuerda, características de cuerpos cavernosos y posición de la uretra3),(16),(20. El tamaño normal del pene de un RN a término debe ser mayor a 2.5 cm y en un RN pretérmino mayor a 2 cm, longitudes menores se consideran micropene3),(24. La longitud normal del clítoris de un RN a término es 0.2 - 0.85 cm, longitudes mayores se consideran clitoromegalia3),(24.
Gónadas: si se palpa la presencia de gónadas en bolsa escrotal o en canal inguinal lo más probable es que sea un testículo o en casos raros un ovotestes8),(20. El tamaño normal del testículo en un neonato a término es 1.1 mL3.
Pliegues labioescrotales, periné y sacro: Se debe realizar una inspección y palpación cuidadosa, mencionando características como rugosidad, pigmentación, fusión y simetría1),(2),(16),(21. Se debe determinar el número de orificios en el periné, considerando la unión de la uretra y la vagina como un seno urogenital1) y por último, descartar malformaciones anorrectales y el sacro ante la posibilidad de anomalías de fusión de arcos vertebrales16.
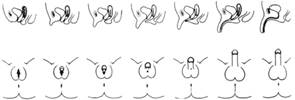
La escala de Prader es comúnmente usada para determinar grado de virilización de los genitales externos en pacientes 46XX. La escala del grado de masculinización se usa en pacientes 46XY y tiene un rango de puntaje entre cero y doce, en donde a mayor puntaje más virilización. (Ver Figuras 2 y 3).

Adicionalmente, se debe hacer búsqueda de anomalías en otros sistemas, considerando la presencia de un síndrome asociado a ADS8. Hay que descartar la presencia de posibles rasgos dismorficos asociados, como por ejemplo defectos en la línea media que sugieran alteraciones en el eje hipotalamo-hipofisis16.
Se debe evaluar presión arterial, frecuencia cardiaca, gasto urinario y signos de deshidratación, que podrían orientar hacia una crisis adrenal con perdida salina8; esta se presenta por deficiencia de mineralocorticoides y ocurre alrededor de la segunda semana de vida, en un 70 % de pacientes con HSC3),(20.
Exámenes e imágenes
Los estudios de primera línea en RN con ADS son básicamente tres, los cuales incluyen:
Estudio hormonal: se debe realizar a partir de las 48 horas de vida, estos incluyen medición de 17-hidroxiprogesterona y delta 4 androstenediona, para descartar la presencia de HSC por déficit de la enzima 21 hidroxilasa7),(17),(24),(25. Adicionalmente, se deben realizar medición de testosterona total, FSH, LH, HAM e inhibina B; las dos últimas permiten evaluar la función de las células de Sertoli16),(26. Con el test dinámico con hCG se puede evaluar la función secretora de andrógenos por las células de Leydig1),(27). En algunos casos se podrá solicitar DHT, cortisol, ACTH basales, entre otros8,24.
Cariotipo de sangre periferia de alta resolución y en algunas oportunidades se solicitará determinación de gen SRY mediante FISH1),(8),(24.
Estudios de imagen como ultrasonido abdominopélvico es útil para la búsqueda de alteraciones de la glándula adrenal, presencia de gónadas y estructuras müllerianas1),(20),(24. También podrá ser necesaria una ecografía renal y de vías urinarias por el origen embrionario común; un ecocardiograma es útil para descartar defectos de línea media o asociados como en la DG o Síndrome de Turner10.
Entre otros estudios complementarios a realizar según lo que se requiera estudiar están:
Cistouretrografía retrograda, muy poco utilizada, permite evaluar la uretra, seno urogenital, tracto vaginal y la detección de fístulas3),(8),(20),(28.
Laparacoscopia, es muy útil para la evaluación objetiva de gónadas no palpables o intraabdominales y el tipo de estructuras o remanentes mullerianos. En algunos casos, permite la realización de biopsia gonadal para llegar a un diagnóstico histopatológico3),(20),(29.
La Resonancia nuclear magnética son alternativas a la laparoscopia, para identificar la localización de gónadas cuando no son visibles por ecografía, pero con una baja especificidad8),(15),(28.
Manejo farmacológico
No hay dudas que en los casos de RN 46XX con ADS secundaria a HSC, se deben hacer suplencias hormonales con glucocorticoides y/o mineralocorticoides con el objetivo de generar un balance entre la producción de andrógenos y cortisol; de modo que no se produzca una detención en el crecimiento, una aceleración de la mineralización ósea o una alteración mayor en sus genitales externos1),(30. En la mayoría de los casos, la supresión de la secreción androgénica suprarrenal permite una regresión en el tamaño del clítoris1.
En los casos de anomalías gonadales con defectos en la síntesis de andrógenos, si el grupo transdisciplinario decide conducir a una asignación masculina se debe hacer suplencia con enantato cipionato de testosterona1),(10),(20; los pacientes con PAIS deben recibir dosis mayores de testosterona para una adecuada virilización23. El crecimiento del pene en respuesta al tratamiento es variable y depende de la condición subyacente1.
Manejo quirúrgico
Es un tema muy controversial que debe ser discutido por el equipo transdisciplinario, en donde se tendrá en cuenta la edad y sexo asignado, los riesgos, beneficios y efectos secundarios del procedimiento.
Estas intervenciones deben realizarse por cirujanos especializados en centros con elevada experiencia y cumplir los siguientes objetivos: 1) Conseguir una apariencia genital compatible con el sexo asignado; 2) Lograr un aspecto estético satisfactorio; 3) Permitir una función sexual y reproductiva optima; 4) Reducir riesgo de infección, incontinencia u obstrucción urinaria; 5) Reducir el riesgo de malignidad gonadal1),(20. En todo caso, siempre se privilegia el resultado funcional sobre la apariencia estrictamente cosmética, por lo que es importante informar a los padres y al paciente para no crear falsas expectativas en torno al resultado2),(20.
Extirpación de gónadas y riesgo tumoral
La decisión de realizar gonadectomía debe contemplar factores como riesgo elevado de malignización gonadal, potencial de fertilidad y funcional gonadal y cuando el género asignado es diferente al sexo gonadal3),(8),(31.
Los tumores germinales son los más frecuentes en las ADS. Su forma invasiva es el seminoma en el testículo y el disgerminoma en el ovario1),(16),(31). Se consideran factores de riesgo de malignización como: la edad (a mayor edad mayor riesgo), la localización gonadal (mayor riesgo en gónadas intraabdominales), el diagnóstico (mayor riesgo en PAIS que en CAIS), la presencia de gen SRY en una paciente 46XX y, fundamentalmente, las gónadas disgenéticas que tienen mayor riesgo de malignidad10),(20),(31.
Manejo transdisciplinario
El manejo de estos pacientes debe ser realizado en un centro de referencia que incluya un grupo de especialistas con experiencia en el tema como endocrinólogo pediatra, genetista, urólogo pediatra, cirujano pediatra, ginecólogo, neonatologo, radiólogo, trabajadora social, psicólogo e idealmente un especialista en ética médica2),(3),(12.
Asignación de sexo
El médico que realiza la primera evaluación no está obligado a asignar un sexo en el certificado de nacimiento para evitar falsas expectativas en la familia sobre la asignación y por otro lado, evitar dificultades a nivel legal y psicosocial.
Los factores que influyen en las decisiones para la asignación de sexo incluyen: el diagnóstico, la anatomía de los genitales internos y externos, el grado de función gonadal, el potencial de fertilidad, la capacidad de respuesta a la terapia hormonal, sexo cromosómico y las opciones quirúrgicas y los resultados; adicionalmente, factores como deseos de la familia, creencias culturales o sociales, también deberían tenerse en cuenta16), (20),(32.
Enfoque integral
Considerar dentro del manejo todas las necesidades que puedan tener incluyendo el estado emocional, social y económico33),(34. Siempre brindar una comunicación efectiva entre el grupo médico, los familiares y el paciente, aportando información clara y precisa acerca de la condición actual y a futuro3),(10. Es necesario abordar las preocupaciones y dudas que se generen en cada etapa del desarrollo del paciente, temas que deberán ser asesorados por expertos en salud mental2,16.
Consentimiento informado
Diligenciar el consentimiento informado a todos los pacientes con ADS, especialmente cuando se va a realizar un examen físico detallado y/o toma de fotografías20),(35. Es necesario mantener la confidencialidad y no se deberá hacer divulgación de información sin previa autorización de un representante legal10),(35.
Conclusiones
Las ADS es un tema poco conocido por el personal de salud, si bien, la prevalencia de RN con genitales ambiguos no es tan elevada, en la práctica clínica sí puede ser común evaluar pacientes con alteraciones genitales menores que deberán ser estudiados.
Es necesario que todos los profesionales relacionados con estos pacientes empleen los términos adecuados al momento de realizar el examen físico genital, así mismo, deben usar la nueva nomenclatura en el diagnóstico de las ADS.
La causa más común de genitales ambiguos es la HSC, por lo que la importancia de este tema es reconocer la urgencia endocrina en caso de presentarse una crisis adrenal que pueda poner en riesgo la vida del paciente.
Adicionalmente, los equipos transdisciplinarios son esenciales para el abordaje diagnóstico y terapéutico de esta compleja condición; temas como asignación de sexo e indicación quirúrgica pueden llegar a preocupar a la familia y es necesaria la presencia de un grupo médico con experiencia en el tema.

