











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La leucemia es el nombre común atribuido a varios trastornos neoplásicos malignos que afectan el tejido hematopoyético. Dichas enfermedades se caracterizan por una proliferación clonal anormal de precursores celulares, que invaden de forma difusa la medula ósea. Esta infiltración, conlleva a fallas en la función medular normal, que se traducirán en la presentación clínica característica de la enfermedad. Las células neoplásicas, a su vez, pueden escapar a sangre periférica, posibilitando así la infiltración de otros órganos no hematopoyéticos 1), (2.
Las leucemias agudas se presentan tanto en niños como en adultos. En población pediátrica es el tipo de cáncer con mayor tasa de incidencia. En población adulta, aunque esta enfermedad es menos frecuente, presenta un comportamiento altamente agresivo con una remisión completa (RC) del 75% y una supervivencia libre de evento (SLE) a largo plazo que no supera el 30% 3), (4), (5), (6), (7. En general, las neoplasias hematológicas representan la undécima y décima causa más frecuente de aparición y muerte por cáncer en todo el mundo, respectivamente, con más de 350.000 casos nuevos diagnosticados y 265.000 muertes al año 8. Colombia es el segundo país de Latinoamérica con mayor incidencia de leucemia, con una incidencia promedio de 5.8 por 100.000 habitantes, panorama que se acompaña de un pobre desenlace clínico, ocupando el octavo lugar entre las muertes por cáncer en el país, con una tasa de mortalidad ajustada por edad del 4,0 y 3,2 por 100.000 habitantes en hombres y mujeres, respectivamente 9.
Adicionalmente, la supervivencia de pacientes con leucemia en Colombia es más baja que en otros países del mundo 5.
Una de las principales causas de la baja supervivencia en pacientes con leucemia es la resistencia al tratamiento que se caracteriza por la presencia de células tumorales después de la quimioterapia, lo que lleva al establecimiento de la enfermedad mínima residual (EMR). El desarrollo de resistencia a los medicamentos durante la quimioterapia, conlleva a fracasos del tratamiento y a la enfermedad recurrente, lo que se traduce en un mal pronóstico de la enfermedad 10. Por lo anterior, el estudio de los mecanismos de resistencia a la quimioterapia es un objetivo primordial de investigación en cáncer, pues su entendimiento permitirá el desarrollo de esquemas terapéuticos efectivos. En este contexto, esta revisión pretende abordar los avances más recientes sobre los mecanismos involucrados en la resistencia tumoral en leucemia.
Quimioresistencia tumoral
El desarrollo de resistencia a la quimioterapia en pacientes con leucemia, y en general en cáncer, es un fenómeno multifactorial, en el cual pueden intervenir tanto factores farmacológicos como celulares. La farmacocinética de los agentes quimioterapéuticos, es decir, su distribución en el organismo, mecanismo de acción o activación, así como la presencia de barreras fisiológicas como la infiltración del sistema nervioso central por células tumorales, pueden limitar la biodisponibilidad de los fármacos en el sitio activo del proceso tumoral. Este tipo de resistencia, conocido como resistencia farmacológica, puede ser modulada al ajustar la vía de administración, dosis o el esquema de administración del fármaco 11), (12), (13), (14.
En cuanto a los factores celulares, hasta hace algunas décadas, la resistencia al tratamiento farmacológico se asociaba únicamente a la sobre-expresión de la glucoproteína de permeabilidad (P-gp), una ATPasa transmembranal presente en la superficie celular, codificada por el gen MDR1 (Multi Drugs Resistance 1, por sus siglas en inglés), ahora conocida como un miembro de la superfamilia de transportadores ATPbinding cassette (ABC). Aunque la P-gp se encuentra ampliamente distribuida en el organismo y participa en el transporte de diversas moléculas a través de membranas extra e intracelulares; se sabe que en las células tumorales, es responsable de la extrusión activa de diferentes tipos de agentes quimioterapéuticos al medio extracelular, con la consiguiente reducción de la concentración intracelular de éstos, lo que imposibilita su acción citotóxica 15), (16.
Sin embargo, recientes estudios han aumentado nuestro conocimiento sobre los procesos biológicos que regulan la supervivencia y la muerte en células tumorales, permitiendo identificar otros mecanismos asociados con la quimiorresistencia de células tumorales, entre los cuales se encuentran: mecanismos de eliminación de fármacos por sobreexpresión de transportadores celulares no asociados a P-gp 17; mecanismos relacionados con la maquinaria de apoptosis celular 18), (19; alteración en los mecanismos de desintoxicación, mejoras en los procesos de reparación del DNA, cambios en la regulación del ciclo celular, así como la regulación negativa de los blancos biológicos de medicamentos 13), (20), (21. No obstante, adicional a los mecanismos de resistencia anteriormente expuestos, durante los últimos años dos teorías han venido tomando fuerza como posible explicación de la quimiorresistencia desarrollada por los pacientes con leucemia: la teoría de las células madre de cáncer como posibles responsables de la recurrencia de la enfermedad 22, y los mecanismos de evasión de muerte celular a través del desarrollo de estrategias metabólicas encaminadas a la modulación de la función mitocondrial, por alteraciones en el equilibrio energético celular 12), (23.
Teoría de Células Madre Leucémicas
En el sistema hematopoyético, así como en otros tejidos, las células madres normales (Hematopoietic Stem Cell, HSCs por sus siglas en inglés) poseen tres características que las hacen únicas: la capacidad de auto-renovación, capacidad de diferenciación en múltiples linajes (pluripotencia) y su extenso potencial de proliferación. Aunque las células madre pueden renovarse por sí mismas, generalmente son inactivas, y pasan la mayor parte de su tiempo en G0/G1.
En los últimos 30 años ha habido un creciente interés en la identificación de la población maligna responsable de la formación y mantenimiento del tumor así como el inicio de la recaída. Células con cualidades de células madre han sido identificas tanto en tumores malignos de origen hematopoyético, como en algunos tumores sólidos, demostrándose además que dicha población tiene la capacidad de inducir la enfermedad de novo 24. Se han empleado varios nombres para identificar a esta población, siendo el término más aceptado el de Células Madre de Cáncer (Cancer Stem Cells, CSCs por sus siglas en ingles) y específicamente para el caso de las leucemias, esta población ha sido llamada Células Madre Leucémicas (Leukemia Stem Cells, LSCs por sus siglas en inglés), ya que existen en el ápice de una organización jerárquica y por analogía a las células normales primitivas del sistema hematopoyético, poseen atributos funcionales que incluyen: la capacidad de iniciar, mantener y propagar en serie la leucemia in vivo mientras retienen la capacidad de diferenciarse en la progenie comprometida que carece de estas propiedades 25.
La existencia de las células madre de cáncer fue postulada hace más de 50 años por Bruce WR, et al. 26, pero fue probada solo hasta la década de los 90´s por Lapidot, et al. 27 en 1994 y más tarde por Bonnet y Dick 25) en 1997, quienes identificaron una pequeña subpoblación de células CD34+/CD38- de leucemia mieloide aguda humana capaz de iniciar el proceso leucémico en trasplantes seriales en un modelo murino NOD/SCID los cuales fueron histológicamente similares a los del donante, mientras que la otra población de células madre del tumor que eran CD34+/ CD38+ no poseía esta capacidad. También se demostró que la frecuencia de LSCs en el tumor podía variar entre 1 en 10.0000 células a 1 en 100’000.000 células. La identificación de LSCs en malignidades hematológicas condujo a la confirmación de las CSCs en numerosos tumores sólidos presentes en órganos como cerebro, colon y mama 28.

Hoy en día, la evidencia científica demuestra que la evolución molecular de las LSCs (Figura 1) inicia con alteraciones genéticas a nivel de la célula madre hematopoyética como la traslocacion BCR-ABL en el caso de algunos subtipos de leucemias, lo que da como consecuencia la expansión de poblaciones progenitoras multipotentes y progenitores específicos de linaje 29. La adquisición de anormalidades adicionales en la transducción de señales confiere a las LSCs un aumento de la supervivencia y capacidad de autorenovación. Estas alteraciones celulares intrínsecas junto con las alteraciones derivadas de las señales extracelulares provenientes del microambiente favorecen la expansión y la supervivencia de las LSCs así como la subsecuente recaída de la enfermedad y la resistencia a las terapias estándares. Las LSCs comparten muchas características con sus análogas normales, incluyendo su estado quiescente, resistencia a la apoptosis y capacidad de autorenovación. Sin embargo, las LSCs poseen además características que habilitan su capacidad de supervivencia a largo plazo y su escape de los efectos citotóxicos de la quimioterapia como la expresión de bombas de eflujo multiresistentes (P-gp) 10), (15), (17, lo cual reduce el estrés citotóxico al cual son sometidas durante la quimioterapia y permite que las LSCs se conviertan en un reservorio para la selección de mutantes resistentes a la terapia dirigida o convencional 30. Las LSCs se caracterizan también por una capacidad de autorenovación ilimitada gracias al predominio de vías clave de señalización conservadas en el desarrollo, como Bmi-1 31, Wnt/β-catenina 32), (33 y Hedgehog 34. Además, las LSCs pueden evadir la respuesta inmune por medio de la sobreexpresión de CD47, una proteína que une diferentes ligandos, entre ellos la proteína reguladora de señales (SIRP-alfa), presente en células mieloides, interacción que conduce a una señalización bidireccional, resultando en diferentes respuestas celulares incluida la inhibición de la fagocitosis, lo que impide su destrucción por los macrófagos 35. Adicionalmente, se ha descrito que las LSCs tienen un ciclo celular quiescente 36), (37 lo cual explicaría por qué los esquemas convencionales anti proliferativos no son efectivos en esta población celular.
Las células madre hematopoyéticas (HSC) dan lugar a células progenitoras, células progenitoras comprometidas con linajes hematopoyéticos específicos y finalmente a células diferenciadas maduras. Los eventos oncogénicos en HSC o células progenitoras producen una población de células madre con autorenovación aberrante lo cual resulta en una lesión preleucémica que progresa a una transformación maligna. En el momento del diagnóstico, el tumor heterogéneo contiene tanto células normales como células madre leucémicas (LSC) que tienen la capacidad de auto renovarse y de dar origen a células leucémicas diferenciadas. A medida que la enfermedad progresa, la infiltración leucémica es mayor. Las mutaciones adicionales permiten que las LSC adquieran un fenotipo resistente mediante el aumento de las vías de señalización de proliferación, el aumento de los mecanismos antiapoptóticos y la evasión de la respuesta inmune. Después del tratamiento quimioterapéutico, las LSC pueden evadir la muerte y sobrevivir a las terapias gracias a las propiedades adquiridas, lo que finalmente lleva al paciente a la recaída de la enfermedad.
Evidencias recientes demuestran además que el aumento de activación aberrante de vías antiapoptóticas como Bcl-2 y PI3K/AKT/mTOR, la elevada actividad del factor NF-κB, y la sobreexpresión de vías de señalización implicadas en la auto-renovación NOTCH, así como alteraciones epigenéticas como el aumento de la actividad de enzimas como las histonas deacetilasas que regulan la expresión del DNA por modificaciones de histonas en la cromatina, son factores intrínsecos claves que provocan la persistencia y resistencia de las LSCs 33), (38), (39), (40), (41 así como el aumento en la actividad de la enzima telomerasa 42, mientras que factores extrínsecos que promueven el mantenimiento de las LSCs pueden estar involucrados en la activación de vías como la señalización de CXCR4/CXCL12 que promueve la quimiotaxis y la migración celular hacia el nicho hematopoyético protegido por otras células derivadas del microambiente de la médula ósea como las MSCs (por sus siglas en inglés, Mesenchymal Stem Cells) 43. Estas vías de señalización alteradas son prometedores blancos moleculares para la erradicación selectiva de las LSCs. Inhibidores farmacológicos y biológicos de estas vías de señalización podrían complementar los esquemas actuales de quimioterapia (Figura 2).
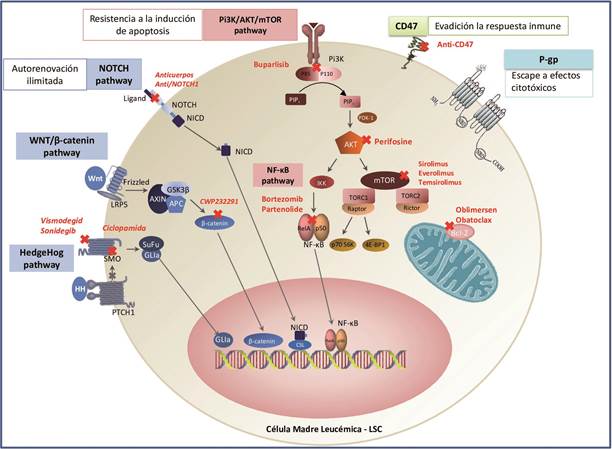
Diagrama de una Célula Madre Leucémica con expresión de diferentes moléculas de superficie como CD44, CXCR4, CD117, VLA-4, TIE-2, CD33, CD123 y activación de vías de señalización que confieren capacidad de resistencia tras ocurrir las mutaciones y eventos leucemogénicos. Las vías de señalización que aumentan la supervivencia como PI3K/AKT y NFκB, pueden activarse en LSCs por diversos estimulos como citokinas, factores de crecimiento y quimiocinas. El potencial de autorrenovación es inducido por la activación de otras vías como HedgeHog, Wnt/Bcatenina y NOTCH1. La sobreexpresión de proteínas antiapoptóticas de la familia Bcl-2 como Bcl-2, Bcl-xl, Bcl-w y Mcl-1 induce la resistencia celular al estrés provocado por apoptosis y disminuyen la acción de proteínas pro-apoptóticas como Bad, Bax y Bim. Las estrategias para la erradicacion selectiva de las LSCs (marcadas en rojo) incluyen antagonistas de citoquinas y quimiocinas, anticuerpos monoclonales dirigidos contra marcadores de superficie expresados por las LSCs y moléculas inhibidoras de diversos componentes de las vías de señalización implicadas en autorrenovación, supervivencia, evasión de apoptosis y metabolismo tumoral.
Debido a que las células madre leucémicas posee un rol decisivo en la formación, desarrollo y crecimiento de la leucemia, esta población celular se ha planteado como posible responsable de la recurrencia de la enfermedad y la resistencia a la quimioterapia convencional observada en gran número de pacientes. Esto ha estimulado el estudio de nuevas estrategias para el desarrollo de terapias contra el cáncer, ya que estas células juegan un papel fundamental en al menos tres diferentes contextos de la progresión del cáncer: 1) Durante la formación del tumor primario, en donde mutaciones en una célula madre normal pueden dar origen a una LSC que a su vez da origen al tumor; 2) durante la recaída de la enfermedad, dado que el tratamiento con quimioterapia destruye la mayoría de las células del tumor primario, pero las LSCs son resistentes a la terapia y el tumor crece de nuevo; 3) y finalmente durante la propagación, ya que las CSCs derivadas del tumor primario pueden migrar a sitios distantes y originar metástasis44 (Figura 3).
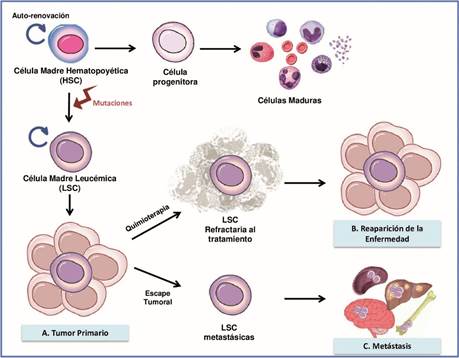
La existencia de LSCs y su relación biológica con mecanismos de resistencia y capacidad de metastasis ha generado nuevos interrogantes y dificultades en el tratamiento de pacientes con Leucemia. Por este motivo se ha estudiado el rol de las LSCs en la recaída y en la enfermedad primaria refractaria. En este contexto, un estudio realizado en pacientes con leucemia mieloide aguda encontró que la persistencia de la población de LSCs dentro del compartimento de células CD34+/ CD38- en pacientes que mostraron una respuesta completa al tratamiento, afectaba la supervivencia libre de enfermedad (5,6 meses en los pacientes con mayor porcentaje de LSCs vs. 16 meses en los de menor porcentaje) y su frecuencia se asociaba con presencia de enfermedad mínima residual, por lo que estos dos factores podrían aportar gran información pronostica 45. Otros estudios, han permitido identificar además un perfil génico diferencial de las LSCs asociado a menores tasas de supervivencia libre de recaída46. Goldman, et al. aislaron y cultivaron células progenitoras CD34+/CD38- y BCR/ABL+ provenientes de pacientes con LMC planteando la posibilidad de la persistencia de una población de LSCs resistentes al tratamiento con inhibidores de tirosin kinasa 47. Corbin, et al. además encontraron que la población de LSCs en LMC no depende de la actividad de BCR-ABL para su supervivencia, hecho que las hace intrínsecamente resistentes a la actividad de los inhibidores de tirosin kinasa 48. George, et al. demostraron la presencia de SL-IC (leukemia initiating cells) en blastos de LLA de sangre periférica de pacientes Philadelphia + con fenotipo CD34+/CD38- con habilidad para iniciar LLA Ph+ en modelos de ratones inmunosuprimidos y con características de proliferación, diferenciación y auto renovación previamente mencionadas en modelos de LMA 49. Otros estudios han sugerido que esta población de LSCs son iniciadoras de la clona tumoral y albergan mecanismos de resistencia a medicamentos. La anterior evidencia permitiría que el desarrollo de estrategias terapéuticas dirigidas específicamente contra las LSCs, puedan erradicar el tumor más efectivamente que los tratamientos actuales y reducir el riesgo de recaída y metástasis.
El metabolismo celular y su rol en la resistencia a la quimioterapia
La reprogramación metabólica de las células tumorales ha sido incluida recientemente al grupo de “características distintivas del cáncer”, un conjunto de propiedades centrales que comparten todas las células tumorales 50), (51), (52), (53. Se ha descrito que las células tumorales presentan un metabolismo celular alterado a través de múltiples mecanismos moleculares que convergen con el fin de proporcionar soporte para suplir las tres necesidades básicas de las células en división: generación rápida de ATP para mantener el estado energético y generar la energía suficiente para soportar la rápida división celular; aumento de la biosíntesis de macromoléculas; y el mantenimiento del estado redox celular 54. De esta manera, las células tumorales reprograman su metabolismo para satisfacer las necesidades previamente mencionadas durante la progresión tumoral. Podría decirse que el mejor caracterizado de estos cambios metabólicos es el efecto Warburg, que hace referencia a la tendencia de las células tumorales a depender de la glicólisis para obtener energía y metabolismo anabólico, incluso cuando los niveles de oxígeno son adecuados para la fosforilación oxidativa 53), (55), (56. Este metabolismo aberrante de la glucosa, conocido también como glicólisis aeróbica, fue descrito por primera vez por Otto Warburg en 1924, y puede entenderse fácilmente cuando se considera que el crecimiento celular depende de la biosíntesis de precursores que se obtienen a partir de intermediarios metabólicos, proporcionados principalmente por las vías glicolítica y el ciclo del ácido tricarboxílico (tricarboxylic acid cycle, TCA por sus siglas en inglés) 57), (58. En los cánceres y los tejidos normales de rápido crecimiento, estas vías son activadas gracias a una mayor absorción celular de glucosa y glutamina, lo cual puede ser visualizado clínicamente mediante la exploración por tomografía por emisión de positrones (PET).
Aunque durante mucho tiempo se creyó que esta anomalía metabólica se debía a un daño mitocondrial irreversible, estudios recientes plantean que es probable que las mitocondrias en el cáncer se supriman activamente 23. Dado que las mitocondrias se integran ampliamente en la biología metabólica, de señalización y apoptosis celular, la supresión mitocondrial en combinación con el metabolismo impulsado por la glicólisis podría conllevar a mecanismos de evasión de la muerte celular intrínseca y promover una rápida proliferación tumoral 55.
Hoy se sabe que muchos estímulos oncogénicos afectan los circuitos metabólicos que promueven el metabolismo de Warburg. Entre estos se encuentra la vía de señalización Notch, que se haya sobre expresada en la mayoría de las leucemias linfoblásticas agudas de células T humanas (T-cell acute lymphoblastic leukaemia, T-ALL por sus siglas en ingles), generalmente como resultado de mutaciones en NOTCH1 59, y se ha visto que promueve el metabolismo glicolítico 60. Trabajos complementarios en esta línea demuestran que Notch además promueve el metabolismo oxidativo; al respecto, Herranz, et al. observaron que la inhibición farmacológica o la ablación genética de la señalización de Notch en modelos murinos xenotransplantados con T-ALL humana primaria, se traducía en un incremento dramático de los niveles de glutamina, mientras que la expresión de enzimas que normalmente aumentan la glutaminólisis (por ejemplo, la isoforma M2 de la piruvato quinasa PKM2 y la glutaminasa GLS) rescataban a los linfoblastos de la inhibición de Notch. Estos resultados demuestran que las T-ALL con mutaciones activadoras de NOTCH1 usan la glutamina como principal fuente de intermedios del ciclo TCA (proceso conocido como anaplerosis), en otras palabras, que Notch influye en el metabolismo intermediario de las células de T-ALL. Por otro lado, Herranz, et al. observaron que la inhibición de la señalización de NOTCH1 de la mano con la inhibición de la glutaminólisis conllevaba a un bloqueo metabólico, el cual desencadenaba un proceso de autofagia en las células leucémicas. Es decir que, el empleo de inhibidores de la glutaminolisis potenciaba de manera fuerte y sinérgica los efectos antileucémicos de la terapia anti-NOTCH1 en este modelo murino de T-ALL. Estos hallazgos validan el empleo de estrategias terapéuticas enfocadas en bloquear rutas que promueven el metabolismo de Warburg en esta enfermedad61. El vínculo entre Notch y el metabolismo de la glutamina en T-ALL está en línea con el trabajo previo de Palomero y col que evidencia como un objetivo crítico posterior de NOTCH1 en este contexto celular es MYC, un regulador clave de la glutaminólisis 62), (63.
A pesar de que las T-ALL emplean la glutaminólisis como fuente de intermediarios del TCA, se ha observado que algunas T-ALL que presentan sobre expresión de Notch continúan creciendo normalmente a pesar del empleo de anti-NOTCH1. Sobre el tema, Herranz, et al. proporcionan datos convincentes que indican que un modo de escape metabólico a la inhibición de Notch es la regulación positiva de la señalización de PI3K/AKT que conlleva a una mayor dependencia celular de la glicólisis (en lugar de la glutaminólisis) para la anaplerosis 61. Esta regulación positiva de la señalización de PI3K/AKT se logró mediante la pérdida del gen supresor tumoral PTEN, que codifica una fosfatasa lipídica con actividad de freno en la señalización de PI3K/AKT. La pérdida de la función PTEN se ha asociado además con cambios en la expresión de genes que permiten la utilización de la glucosa. Las mutaciones de pérdida de función de PTEN se encuentran en aproximadamente el 20% de la T-ALL humana 60.
El metabolismo alterado de la glucosa se ha descrito como una causa de quimiorresistencia en múltiples tipos de tumores. Un estudio en el que se evaluó el metabolismo celular de pacientes con leucemia mieloide aguda (Acute mieloide leukemia, AML por sus siglas en inglés) resistente a los medicamentos, demostró que el aumento de la expresión de genes asociados a la vía glicolítica, incluido el factor inducible por hipoxia 1α (HIF-1α), el transportador de glucosa 1 (GLUT1), la hexoquinasa-II (HK-II) y la lactato deshidrogenasa (LDH) están asociados con la resistencia a los medicamentos en células de AML. También se observó que la inhibición de la glucólisis mediante el uso de inhibidores glicolíticos hacía que las células de AML resistentes fueran susceptibles a la doxorrubicina, un fármaco ampliamente utilizado en la quimioterapia del cáncer 64. Otros autores han asociado la resistencia quimioterapéutica en leucemia con el metabolismo alterado de la glucosa: la resistencia a la prednisolona en pacientes con leucemia linfoblástica aguda de precursores de células B (B cell acute lymphoblastic leukaemia, B-ALL por sus siglas en inglés) se asoció con una mayor expresión de genes implicados en el metabolismo de la glucosa 65), (66; en células de leucemia mieloide crónica (chronic myeloid leukemia, CML por sus siglas en inglés) resistentes al imatinib también se encontró exhibían un fenotipo de glucólisis alta, con una elevada absorción de glucosa y producción de lactato en comparación con las células sensibles al imatinib 67), (68. Además, las líneas celulares de AML con un mayor nivel de glucólisis mostraron una mayor resistencia a la inducción de apoptosis in vitro 69.
El metabolismo de la glutamina también se ha vinculado con desarrollo de quimiorresistencia en leucemia, estudios recientes observaron un incremento robusto en la expresión de glutamina amoniaco ligasa (GLUL) y del contenido celular de glutamina posterior al tratamiento con glucocorticoides, hormonas metabólicas con efectos inmunosupresores que han demostrado ser medicamentos eficaces contra la leucemia linfoblástica aguda (LLA) infantil. Esta síntesis aumentada de glutamina redujo el contenido de autofagosoma de las células leucémicas y rescató la viabilidad celular 12. Estos datos sugieren que la síntesis de glutamina afecta la autofagia y posiblemente el inicio de la muerte celular en respuesta a los GC, pudiendo conferir una mayor resistencia a la quimioterapia, tal como se ha observado en pacientes y en líneas celulares de leucemia 12.
Aunque el efecto Warburg es ampliamente ventajoso para el crecimiento de muchas células tumorales, no todas las células de cáncer exhiben este metabolismo glicolítico, fenómeno conocido como “flexibilidad o plasticidad metabólica”. Esta variabilidad se ha atribuido en parte a la privación de oxígeno y nutrientes en los tumores sólidos donde con frecuencia se produce necrosis central y pobre vascularización, pero tales factores anatómicos no están tan claramente involucrados en las leucemias, que residen en la médula ósea y tienen fácil acceso a la vasculatura. Estudios en esta línea han determinado que las células tumorales pueden emplear fuentes alternativas de energía a través de la descomposición de ácidos grasos, aminoácidos y lactato, o preferencialmente por Fosforilación oxidativa mitocondrial (OXPHOS) 53), (70), (71. Un ejemplo de estos son los resultados obtenidos por Goto, et al. quienes evaluaron el crecimiento y metabolismo energético de líneas celulares de leucemia bajo condiciones de hipoxia, esto considerando que al igual que las HSCs normales, las células leucémicas proliferan en la médula ósea donde el suministro de oxígeno es limitado; demostrando que las células de leucemia no siempre dependen de la glicólisis para la producción de energía, sino que pueden migrar su metabolismo a OXPHOS, mostrando la capacidad de adaptación de estas células 72.
Por su parte, Kishton, et al. encontraron que a diferencia de PI3K, la quinasa activada por AMP 5’ (AMPK) puede inhibir la señalización de mTORC1, esto con el objetivo de contrarrestar las señales oncogénicas múltiples, incluidas las de Ras y Myc, que conllevan al desarrollo de estrés metabólico crónico en las células leucémicas71. En este escenario, la AMPK es activada y suprime la señalización de mTORC1 y la glicólisis, mientras se apoya en el metabolismo mitocondrial (OXPHOS) con el fin de asegurar la supervivencia de las células T-ALL in vivo. Estos resultados demuestran que las células leucémicas utilizan un programa metabólico flexible en el que el estrés metabólico inducido por Notch conduce a la activación de AMPK, el cual desempeña un doble papel: inhibir el metabolismo anabólico y promover la supervivencia de las células T-ALL a través de la regulación positiva del metabolismo mitocondrial. Otros autores han informado que AMPK puede ser importante en la mitigación del estrés metabólico al que son sometidas las células que inician la leucemia mieloide73 y en células T activadas in vivo 74.
Así mismo, estudios sobre el estado oxidativo y propiedades bioenergéticas de células madre leucémicas en muestras de pacientes con leucemia mieloide aguda (Acute myeloid leukemia, AML por sus siglas en inglés) evidenciaron que la población tumoral responsable de la resistencia y recaída consistía en una población más quiescente y con actividad metabólica reducida presentando menor metabolismo tanto oxidativo como glicolítico en comparación con las células tumorales sin características de células madre 19. De igual forma, estudios realizados en T-ALL observaron que la glicólisis aeróbica se encuentra menos activa en las células de LLA de precursores T comparado con células T proliferantes normales y que las células de T-ALL son metabólicamente distintas promoviendo el metabolismo oxidativo y la actividad del complejo I mitocondria 71. Esto sugiere que de una u otra forma, la regulación del metabolismo tumoral, puede jugar un papel en la modulación de la resistencia a la quimioterapia. Lo que hace del metabolismo un tema relevante de estudio para entender el papel del mismo en la resistencia farmacológica y en el desarrollo de nuevas terapias en leucemias agudas.
Es importante resaltar que la teoría metabólica del cáncer que sugiere un estado pro-proliferativo y resistente a la apoptosis, es compatible con la teoría genética del cáncer que ha dominado el campo durante más de cuarenta años pero que para algunos tipos de cáncer no ha logrado ofrecer terapias lo suficientemente eficaces y selectivas, ya que muchos de los cambios oncogénicos que se sabe afectan las vías de señalización intracelular desempeñan un papel activo en la mediación de estos cambios metabólicos, que a su vez, funcionan para apoyar el crecimiento y la replicación de las células tumorales. Por lo tanto, evaluar la asociación entre los programas metabólicos diferenciales y los perfiles genéticos exhibidos por pacientes con leucemia, permitirá identificar posibles objetivos terapéuticos relevantes en esta enfermedad, que pueden tener potencial para traducirse en herramientas terapéuticas efectivas en la detención del crecimiento tumoral y la metástasis.
Perspectivas terapéuticas en evaluación
Con la evidencia existente del rol de las LSCs y el metabolismo de las células leucémicas en procesos como resistencia a medicamentos, falla al tratamiento convencional, recaída y capacidad metastásica, se han planteado diferentes agentes experimentales dirigidos a bloquear los diferentes mecanismos utilizados por las células leucémicas para evadir la acción de los agentes quimioterapéuticos convencionales (Figura 2). En el caso del tratamiento de LSCs, un reto importante en el diseño de nuevos medicamentos dirigidos a controlar esta población tumoral es el que dichas células se asemejan mucho más a las HSCs que a su misma progenie diferenciada, por lo que la potencial toxicidad de las nuevas estrategias terapéuticas debe ser ampliamente estudiada en modelos animales en los cuales se evalúe el injerto y repoblación de células normales, junto con la toxicidad hematológica en estudios clínicos 75.
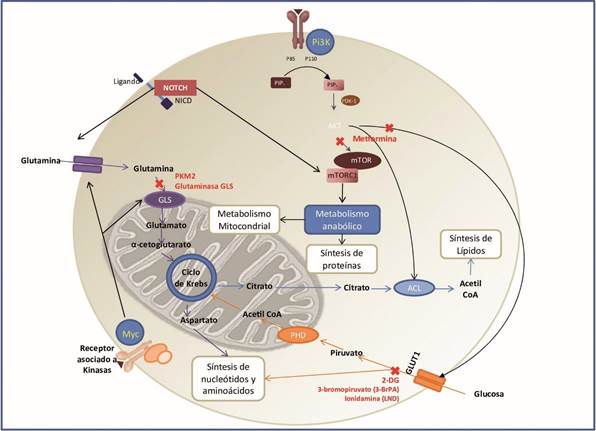
Un número importante de los agentes farmacológicos que se encuentran en la actualidad en evaluación, se dirigen a bloquear vías de señalización y rutas metabólicas sobre expresadas en las LSCs y células leucémicas 76. Algunas investigaciones en las que se evaluó la acción de la Ciclopamida, un antagonista natural de la proteína transmembrana Smoothened (Smo) que desempeña un papel importante en la vía de autorenovación celular HedgeHog activada de manera aberrante en las LSCs 34), (41, demostraron que la ciclopamida induce la apoptosis de células leucémicas CD34+ y disminuye la resistencia a citarabina en modelos animales 77; sin embargo, sus características farmacocinéticas han imposibilitado continuar con su evaluación en la fase de ensayos clínicos 78. Otros inhibidores de la vía HedgeHog como el Vismodegib y Sonidegib, han sido probados en tumores sólidos, y actualmente este último se encuentra en estudios clínicos de fase 1 en pacientes con neoplasias mieloides 79. Diferentes moduladores de la vía Wnt/bcatenina han sido diseñados y aún se encuentran en fases iniciales de estudios clínicos; entre ellos, un inhibidor de CD27, receptor transmembranal miembro de la superfamilia de receptores del factor de necrosis tumoral (TNF) que se encuentra presente en LSCs y células leucémicas y ha demostrado ser un importante activador de la vía Wnt/b-catenina en líneas celulares y modelos animales 80.
También se han diseñado inhibidores de los mecanismos empleados por las LSCs para evadir la apoptosis y mantener su crecimiento aberrante, tales como: el aumento en la actividad del factor NFkB, la alteración en la funcionalidad de componentes de la familia de proteínas Bcl-2 y el aumento en la actividad de la vía Pi3K/AKT/mTOR. En esta línea se han venido evaluando fármacos como el Bortezomib, un inhibidor selectivo y reversible del proteasoma 26S que ha sido utilizado de manera convencional para el tratamiento de mieloma múltiple, el cual ha demostrado tener efectos anti NFkB, y en combinación con la quimioterapia convencional ha mostrado tasas de RC cercanas al 80% en pacientes pediátricos con ALL. También se encuentran en desarrollo estudios clínicos fase II/III de bortezomib en neoplasias mieloides 81), (82. Diferentes inhibidores de Bcl-2 han sido desarrollados como otra alternativa para el tratamiento de leucemia, entre los cuales, aquellos que se encuentran en fases más avanzadas de estudio son el Oblimersen, un oligonucleótido de fosforotioato antisentido, que está siendo probado en pacientes con CLL y el Obatoclax, también conocido como GX15-070, un medicamente experimental que está siendo evaluado en diferentes neoplasias hematolinfoides 83, que han demostrado discreto aumento en la supervivencia libre de progresión. Numerosos agentes que interactúan con diferentes componentes de la vía PI3K/AKT/mTOR se encuentran en ensayos clínicos en la actualidad. Esta vía se considera un blanco terapéutico muy importante pues no solo se ha visto implicada en la resistencia a la apoptosis celular sino que además impulsa el metabolismo anabólico y la tumorigénesis mediante la reprogramación de las mitocondrias.
Dentro de los múltiples fármacos en evaluación, los más prometedores como inhibidores de mTOR son el Sirolimus, everolimus y temsirolimus84; el perifosine como inhibidor de AKT 85; y el buparlisib (BKM120) un pan inhibidor de Pi3K estudiado también en mielo fibrosis y otros tumores sólidos 86.
Por otro lado, teniendo claro que el metabolismo tumoral anormal puede ser un resultado secundario de alteraciones corriente arriba posiblemente influidas por factores genéticos y que la remodelación metabólica del cáncer es necesaria para la supervivencia celular, es posible identificar muchos objetivos terapéuticos. Diversas estrategias que restauran el metabolismo normal han demostrado selectividad y eficacia en estudios preclínicos. El fracaso del tratamiento quimioterapéutico en leucemia ha sido atribuido con frecuencia a la resistencia a los glucocorticoides (por ejemplo, Prednisolona), lo cual se ha asociado directamente con el aumento de la glucólisis en las células tumorales 87. Es por esto que, diferentes compuestos que inhiben específicamente el metabolismo glicolítico están sido evaluados actualmente como alternativas terapéuticas en cáncer 38), (65), (88. Uno de estos es la 2-desoxi-D-glucosa (2DG), un análogo sintético de la glucosa, que se absorbe a través de los transportadores de glucosa (GLUT) y se fosforila mediante la hexoquinasa (HK) para formar 2-DG-6-fosfato (2-DG-6-P). El 2-DG-6-P se acumula dentro de la célula y no se continua metabolizando, lo que induce la detención del crecimiento celular y la muerte celular gracias a la inhibición de dos enzimas glucolíticas: la HK y la fosfoglucosa isomerasa (IGP) 89), (90), (91. Aunque 2-DG ha mostrado resultados promisorios en sistemas celulares y modelos animales, su eficacia como monoterapia está limitada debido al riesgo de toxicidad sistémica a dosis altas. No obstante, el uso de 2-DG en combinación con la terapia convencional ha demostrado que sensibiliza las células leucémicas a los agentes quimioterapéuticos 87), (92, por lo cual podría ser empleado en dosis bajas como coadyuvante del tratamiento en cáncer. Así como la 2-DG, otros inhibidores de la HK, como el 3-bromopiruvato (3BrPA) y la lonidamina (LND) están siendo evaluados en la actualidad como alternativas terapéuticas en ensayos preclínicos y de fase temprana. En el caso del 3-BrPA se ha demostrado que la inhibición de la glicólisis por este compuesto no solo mejora los efectos citotóxicos de la daunorrubicina y la doxorrubicina, sino que además suprime notablemente el crecimiento tumoral cuando se usa con doxorrubicina 93.
Un medicamento que ha venido cobrando interés como factor protector y terapia contra el cáncer es la metformina, un fármaco hipoglicemiante oral prescrito ampliamente en el mundo para el tratamiento de la diabetes tipo 2. Las primeras evidencias que vinculaban la metformina con protección contra el cáncer surgieron de grandes estudios de cohorte. Sin embargo, diferentes estudios se han realizado en células tumorales con el objetivo de establecer su posible mecanismo de acción. Algunos autores han sugerido que el potencial antileucémico de este medicamente puede radicar en dos mecanismos: 1) la disminución de la hiperinsulinemia, limitando la expresión de diversos factores de crecimiento semejantes a la insulina; y 2) el bloqueo de vías de señalización dependientes de AMP, principalmente la vía PI3K/AKT/mTOR 94. Al respecto, Vakana, et al. observaron una disminución en la expresión del transcrito BCR-ABL1 en células leucémicas en presencia de metformina, incluidas aquellas líneas celulares con expresión de la mutación T315I 95. Estos resultados son respaldados por Pan, et al. quienes evaluaron el efecto en diversas líneas celulares de la adición de metformina, demostrando que este medicamento sensibiliza a las células leucémicas a la quimioterapia, mientras que la insulina incrementa la quimiorresistencia 96. Otros modelos en líneas celulares de leucemia han propuesto como mecanismo de acción complementario de metformina la inducción de apoptosis mediante la sobrerregulación de vías de estrés celular y proteínas que interactúan en la apoptosis (disminución de la expresión de IRE1α y CHOP) 97. Recientemente en un estudio piloto realizado en el Hospital General de México se adicionó metformina al régimen institucional de tratamiento de pacientes diagnosticados de novo con ALL, logrando reducir el porcentaje de recaídas tempranas (25% vs 48%) en comparación con el grupo que sólo recibió quimioterapia; la dosis administrada en este ensayo fue de 850 mg vía oral cada 8 horas 98.
Conclusiones
Existe suficiente evidencia que sugiere que la desregulación de las funciones de las células madre junto con el metabolismo tumoral juegan un importante papel en la biología de las leucemias. El concepto de heterogeneidad tumoral y el hecho de que no todas las células dentro del tumor sean capaces de dar inicio y mantener el proceso oncogénico sugiere que la erradicación de las células tumorales maduras no determina en su totalidad la eficacia del tratamiento y esto lleva a pensar en la necesaria erradicación de las células madre leucémicas para mejorar el desenlace de la enfermedad. La evidencia creciente deja de manifiesto propiedades biológicas únicas presentes en las LSCs que les permiten sobrevivir a la terapia y dar inicio a la recaída. Por lo tanto, la identificación de las diferencias entre las células madre normales y las células madre leucémicas ha abierto la posibilidad de definir puntos clave de intervención dirigida específicamente contra la población de LSCs sin afectar la población progenitora normal. Tales terapias dirigidas podrían ser mucho menos tóxicas y más eficaces que los esquemas de tratamiento actuales.
En cuanto al metabolismo, es claro que las células leucémicas exhiben un fenotipo metabólico alterado que les permite adaptarse a mayores demandas metabólicas y cambios ambientales. Dicha flexibilidad metabólica facilita la proliferación de la progenie tumoral, al tiempo que media mecanismos de evasión a la quimioterapia convencional. Es por ello que el concepto de atacar el metabolismo del cáncer ha surgido como un enfoque intrigante para el desarrollo de esquemas terapéuticos mejorados, lo cual será posible únicamente al mejorar nuestra comprensión sobre los mecanismos involucrados en la regulación metabólica de las células leucémicas de los pacientes de nuestra población.

