











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
La modulación de los procesos proinflamatorios en tumores malignos asociados con VPH es compleja, ya que implica respuestas inmunes deficientes que posibilitan la persistencia viral induciendo la progresión de las lesiones neoplásicas en el cuello uterino propiciando un microambiente protumoral1,2. La activación de la respuesta inmune innata representa la primera respuesta del sistema inmunitario contra la infección por VPH y es esencial para iniciar una inmunidad adaptativa de larga duración, específica y efectiva3.
Los IFNs tipo I son citoquinas fundamentales para iniciar una respuesta inmune eficaz contra infecciones virales como el VPH4. El IFN kappa (IFN-k) tiene efectos modulatorios anti-virales, se expresa constitutivamente en los queratinocitos y el IFN épsilon (IFN-ε), posee funciones antivirales únicas en la mucosa reproductiva; estos IFNs pueden ser determinantes en el control de la infección por VPH5-8.
Sin embargo, se ha comprobado que los VPH de Alto Riesgo (VPH-AR) han desarrollado mecanismos muy eficientes de evasión inmunológica9, por lo tanto, es importante comprender cómo la infección por VPH modula los procesos inmunológicos de los queratinocitos infectados al favorecer la propagación viral y determinando el desarrollo del tumor, la resistencia al tratamiento y el pronóstico general de los pacientes con cánceres asociados a VPH10-12.
Este virus oncogénico tiene la capacidad de modular la función de muchas moléculas involucradas en la vía de señalización de los interferones (IFNs) (13, un ejemplo es la desregulación de la familia STAT (Transductor de Señal y Activador de la Transcripción), que participa no sólo en procesos de inmunidad, sino también en la proliferación, apoptosis y diferenciación celular14, esto podría explicar parcialmente por qué una parte de las infecciones por VPH pueden persistir en los hospederos sin iniciar una respuesta inmune efectiva y evolucionar a cáncer.
La terapia exitosa contra el cáncer y la eficaz estratificación individual de los pacientes con cáncer requieren una comprensión completa de la compleja interacción entre las infecciones persistentes con virus oncogénicos, las vías inflamatorias inducidas, el metabolismo de las células cancerosas, los resultados clínicos y la resistencia terapéutica15,16. Hasta ahora, estas conexiones no son bien comprendidas. El objetivo de esta revisión es profundizar sobre las funciones de los IFNs tipo I en la infección causada por el VPH y los mecanismos de evasión de este virus para inactivar los IFNs tipo I.
Generalidades
Los oncovirus a través de sus oncogenes promueven algunas de las características distintivas del cáncer que, sumadas a otros factores como la inmunosupresión o la inflamación crónica, pueden conducir al establecimiento de malignidades. En general, se estima que los oncovirus causan cerca del 12 % de todos los cánceres humanos a nivel mundial, siendo particularmente el VPH el responsable del 5 % de estos cánceres17-19.
El VPH se transmite principalmente a través del contacto sexual y es la infección viral más común del sistema reproductivo20. Aproximadamente, entre el 50 y el 80 % de las personas sexualmente activas se infectan con uno o múltiples genotipos de VPH en algún momento o repetidamente durante sus vidas21. Normalmente, estas infecciones son eliminadas efectivamente por el sistema inmunológico; no obstante, un porcentaje de personas no puede controlar la infección que finalmente resulta en la aparición de lesiones premalignas que pueden progresar a cáncer22. La infección crónica con VPH-AR es la causa del Cáncer de Cuello Uterino (CCU) y se ha asociado a otros cánceres anogenitales, así como a cánceres de cabeza y cuello23.
El CCU es el cuarto cáncer femenino más frecuente a nivel mundial y el segundo más común en mujeres que viven en países de bajos y medianos ingresos, reportándose para el año 2018 un estimado de 570 000 casos de los cuales más de 300 000 tuvieron un desenlace fatal20. En Sudamérica, anualmente hay 40 000 casos nuevos de CCU de los cuáles más de 19 000 resultan mortales24, para el caso de Colombia se estima que hay alrededor de 4 000 casos anuales con casi 2 000 muertes a causa de este tipo de cáncer25. Se ha establecido que la incidencia anual de CCU es de 18,7 casos por cada 100 000 mujeres, siendo la primera causa de muerte por cáncer entre mujeres colombianas de 30 a 59 años, con aproximadamente 5 muertes diarias por esta enfermedad26.
Interferones
Los IFNs fueron descritos por primera vez hace más de 60 años por Isaacs y Lindenmann quienes observaron que los fluidos de los cultivos celulares infectados por virus contenían una proteína celular específica que podía reaccionar con las células para hacerlas resistentes a la infección por virus(27.
Se conocen numerosos subtipos de IFNs en humanos con funciones especialmente implicadas en la respuesta antiviral27-29. Estas funciones son direccionadas por el tipo de receptor, su distribución celular y por las vías de transducción de señales, que una vez activadas promueven la transcripción de más de 2 000 genes estimulados por IFN (ISGs) (30-34. Las proteínas expresadas a partir de estas vías tienen la capacidad de impedir el correcto desarrollo viral: desde la entrada y replicación hasta el ensamblaje y la liberación del virus28,33.
Propiedades y clasificación de los IFNs
Los IFNs son pequeñas glicoproteínas monocatenarias que pertenecen a la familia de las citoquinas. Se componen de 130 a 170 aminoácidos; son activos a bajas concentraciones, tienen acción autocrina y paracrina; son estables en un amplio rango de pH; son menos estables cuando se someten a altas temperaturas y se degradan fácilmente por proteasas, dietil, cloroformo y cetonas35-37. Además, la receptividad al IFN puede variar durante el día, dado que los receptores de IFN exhiben una expresión circadiana, siendo su nivel de expresión mayor en la noche que durante el día38.
Son secretados por células hospedadoras que actúan como sistema inmune innato en la primera línea de defensa contra componentes extraños de tumores, antígenos y patógenos como bacterias, parásitos y virus4. La producción inicial de IFNs no protege directamente, sino que reacciona con receptores específicos en las superficies celulares para activar señales de transducción citoplásmica que luego ingresan al núcleo para estimular genes celulares que codifican una cantidad de proteínas efectoras que conducen a las acciones de defensa4,39.
Antiguamente los IFNs se clasificaban según su origen celular en: IFN-α si eran originados a partir de leucocitos; IFN-β a partir de fibroblastos; e IFN-y a partir de linfocitos T activados. Sin embargo, en 2003 se descubrió el IFN-λ y no se pudo clasificar en ninguna de las categorías preestablecidas40.
Actualmente, los IFNs se clasifican en tres clases dependiendo del receptor a través del cual transmiten una señal intracelularmente: los IFNs tipo I comprendidos por IFNα y sus 13 subtipos, IFN- β, -ε, -k, -t, -δ, -ζ-ω y -v; el IFN tipo II o IFN-y; y los IFNs tipo III que comprenden los IFN-λ1, -λ2 y -λ3 29,41,42.
Señalización intracelular de los IFNs
Los receptores de reconocimiento de patrones (RRP) intracelulares y de superficie celular, como los receptores tipo Toll (TLR) y los receptores tipo RIG-I (RLR), se encargan de reconocer ácidos nucleicos extraños, otros productos microbianos y el ADN propio para inducir los genes que codifican los IFNs tipo I43.
Las funciones de los IFNs son direccionadas por el tipo de receptor, su distribución celular y por las múltiples vías de transducción de señales que una vez activadas promueven la transcripción de más de 2 000 genes estimulados por IFN (ISGs), abarcando un gran espectro de efectos biológicos30-34. La señalización del IFN es específica de la célula y muestra una gran diversidad y complejidad39. Las proteínas expresadas a partir de estas vías tienen la capacidad de impedir el correcto desarrollo viral, desde la entrada y replicación hasta el ensamblaje y la liberación del virus28,33. A continuación, se resumen las principales vías de señalización relacionadas con los IFNs tipo I y se ilustran en la Figura 1.
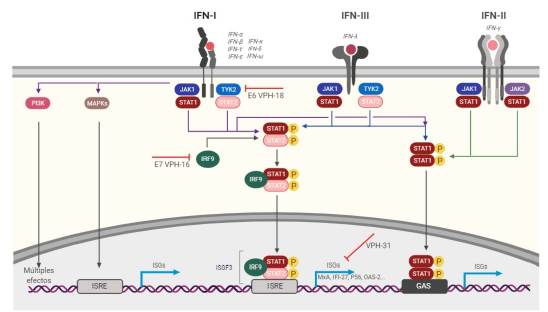
Vía de señalización canónica JAK/STAT
Los IFNs tipo I interactúan y activan los receptores de la superficie celular 1 y 2 (IFNAR1 e IFNAR2). La unión de IFNAR activa las proteínas Janus quinasa 1 (JAK1) y tirosina quinasa 2 (TYK2), estas proteínas se autofosforilan y fosforilan al receptor conduciendo a la dimerización de las proteínas citoplasmáticas transductoras de señal y activadoras de la transcripción 1 y 2 (STAT-1 y STAT-2) (43.
Posteriormente, los dímeros de STAT se translocan al núcleo y se unen al factor regulador de IFN 9 (IRF9) formando el complejo de Factor 3 de Genes Estimulados por IFN (ISGF3). Este complejo se une a los elementos de respuesta estimulados por IFN (ISRE) que son secuencias consenso de ADN que actúan en cis en los promotores de ISG, lo que finalmente promueve una alta transcripción de los ISGs44.
Transducción de señales no clásica
Los IFN tipo I también pueden señalizar a través de homodímeros de STAT1, pero se asocian más con la vía de señalización mediada por IFN-y. Ellos también pueden activar aguas abajo otros heterodímeros y homodímeros de STAT, incluidos STAT3, STAT4 y STAT5. También se pueden activar otras vías de señalización que no van a depender de la actividad de JAK ni de STAT, por ejemplo, las proteínas quinasas activadas por mitógeno (MAPK) y la ruta de fosfoinositol 3-quinasa (PI3K), lo que conduce a diversos efectos sobre la célula 43,44.
En la unión de los IFN a sus receptores, se pueden inducir múltiples vías de señalización aguas abajo, lo que conduce a una amplia gama de efectos biológicos. El transductor de señal canónico y el activador de la transcripción 1 (STAT1) junto con STAT2 y el factor regulador de IFN 9 (IRF9) forman el complejo de señalización ISGF3. Este se une a los elementos de respuesta estimulados por IFN (ISRE) en los promotores de los genes, induciendo una gran cantidad de genes estimulados por IFN (ISG). Los IFN tipo I también pueden señalizar a través de homodímeros de STAT1 u otras vías de señalización independientes de Janus quinasa (JAK) y STAT: las proteínas quinasas activadas por mitógeno (MAPK) y la ruta de fosfoinositol 3-quinasa (PI3K).
Función de los interferones tipo I
Los IFNs tipo I actúan como puente entre la inmunidad innata y adaptativa. Tienen propiedades antivirales, antiproliferativas, proapoptóticas, antiangiogénicas e inmunoestimuladoras. Además, pueden promover la senescencia e inhibir la función de las células madre en respuesta al acortamiento de los telómeros45-51.
La mayoría de los ISGs codificados al final de la vía de señalización no tienen una acción antiviral directa, pero muchos traducen RRP, los cuales identifican moléculas asociadas a virus y dan inicio a la respuesta inmune innata52. Es decir, los IFNs pueden afectar indirectamente diferentes procesos virales modulando funciones del sistema inmune por medio de la inducción de citoquinas, las cuales van a activar a las células inmunes no infectadas con el fin de poner en funcionamiento vías que no puedan ser inhibidas rápidamente por los virus53. Un ejemplo es aumentar la vigilancia inmunológica regulando positivamente moléculas del complejo mayor de histocompatibilidad clase I, mejorando así la presentación antigénica y activando a los linfocitos T, citotóxicos específicos para reconocer los antígenos virales presentados, estimular la respuesta efectora mediada por células y finalmente conducir a la lisis de las células infectadas54,55.
Algunas de las vías bien caracterizadas de la respuesta inmune inducidas por IFN con potencial actividad antiviral directa incluyen cuatro proteínas: la proteína de 15 kDa estimulada por IFN (ISG15), la proteína Mx, la ribonucleasa L (RNasa-L) y la proteína quinasa R (PKR) (28,56. Sin embargo, hay más proteínas que funcionan como efectores antivirales importantes como ADAR1 y APOBEC; Teterina y Viperina; TRIM19; la exonucleasa ISG20; y las proteínas IFIT28,57. Además, el IFN puede inducir ciertos microARN interferentes con respuestas reguladoras de la infección viral. Estas proteínas van a estar involucradas en diversas funciones que implican la remodelación del citoesqueleto; inducción de la apoptosis; corte, empalme, edición y degradación de ARN viral; regulación en la expresión de proteínas; y modificación postraduccional28,56-58.
La ISG15 es una de las proteínas más importantes que se inducen en las infecciones virales debido a que lleva a cabo diferentes funciones a nivel celular. La ISG15 fue identificada en una vía homóloga a la de la ubiquitina y, al igual que esta, se une de manera covalente a una variedad de blancos proteicos para modular su función en un proceso denominado ISGilación, cuya finalidad es controlar los efectos de la presencia de virus con una maquinaria enzimática que es igualmente regulada por los IFN-I. Sin embargo, la ISGilación no resulta en la degradación de las proteínas marcadas como ocurre en la ubiquitinación, sino que evita la degradación del factor regulador de IFN 3 (IRF3), lo que resulta en una mayor expresión de IFN-β Otra de sus funciones es aumentar o disminuir la afinidad y función de proteínas intracelulares; por ejemplo, mediante esta vía se puede regular positivamente el factor de transcripción NF-kB por medio de la unión y supresión de la actividad enzimática de la proteína fosfatasa 1B (PPM1B). Además, ISG15 tiene funciones extracelulares debido a que se secreta y puede actuar como una citoquina. Existen enzimas que hidrolizan la ISG15, de esta manera se puede revertir la ISGilación28.
Las proteínas Mx son una familia de GTPasas inducidas por los IFN-I, en humanos están comprendidas por las proteínas MxA y MxB, que se expresan en diversos tipos celulares y se ubican junto al retículo endoplásmico liso, lo que permite su participación en la gemación de vesículas, organogénesis y citocinesis. Debido a que están involucradas en el tráfico vesicular pueden capturar elementos virales como las nucleocápsides y de esta manera suprimir la replicación de los virus tanto nucleares como citoplasmáticos28.
La proteína 2'5'-oligoadenilato sintetasa 1 (OAS1) normalmente se encuentra en bajas cantidades en el citoplasma como monómero inactivo a menos que exista una estimulación por parte de los IFN-I. Su función consiste en polimerizar el ATP en oligómeros de adenosina que activan la RNasa-L encargada de degradar los RNAs celulares y virales28.
La PKR se expresa constitutivamente en el núcleo y citoplasma. Gracias a los IFN-I se aumenta la cantidad de esta enzima, la cual en presencia de ARNs virales de doble cadena se activa para interferir en la síntesis de proteínas mediante la fosforilación del factor de iniciación de la traducción eucariótica 2 alfa (EIF2α) (28.
Por otra parte, los genes estimulados por IFN (ISGs), además de afectar directa e indirectamente diferentes etapas del ciclo replicativo viral, pueden producir importantes efectos modulatorios sobre el metabolismo celular59.
En el contexto de la infección por VPH, los queratinocitos han desarrollado mecanismos que le permiten disminuir la propagación de VPH, entre estos, la producción de IFNs que modulan vías metabólicas específicas. Las estrategias de los IFNs incluyen la modulación del flujo de fuentes de carbono exógenas, la inhibición de la biosíntesis del colesterol y el agotamiento de poliaminas, triptófano o sustratos de nucleótidos59.
Virus del Papiloma Humano
Generalidades del virus
VPH son un grupo de oncovirus pequeños icosaédricos, sin envoltura y con un tamaño aproximado de 55 nm de diámetro. Su genoma es de ADN de doble cadena y consta de 8 000 pares de bases, aproximadamente. Contienen ocho o nueve marcos de lectura abierto que dan lugar a las proteínas tempranas: E1, E2, E4, E5, E6 y E7 con funciones reguladoras y a dos proteínas tardías: L1 y L2, con funciones estructurales en la cápside viral60.
Según el análisis de secuencia de ADN, los VPH se dividen en cinco géneros: alfa, beta, gamma, mu y nu. Aquellos que se asocian con el desarrollo de cáncer son los alfa-papilomavirus; estos se han clasificado según su tropismo en tipos cutáneos y mucosos. A su vez, los tipos mucosos, dependiendo de su grado de asociación con el cáncer, pueden catalogarse como de alto o bajo riesgo. Los VPH más estudiados son los alfa-papilomavirus mucosos de alto riesgo que causan CCU23.
Ciclo viral
El ciclo del VPH está relacionado con el crecimiento y la diferenciación de los queratinocitos de la capa basal del cuello uterino. Estas células se infectan con VPH a través de lesiones y microabrasiones del tejido. La internalización infecciosa tarda varias horas, después el ADN viral se libera de la cápside y se transporta al núcleo como material genético libre o episomas extracromosómicos. La expresión génica temprana está estrechamente controlada en las células epiteliales basales con una amplificación importante del ADN viral. La replicación se produce sólo en células suprabasales que se diferencian, siendo destinadas a la madurez y la senescencia; por lo tanto, no expresan naturalmente la maquinaria replicativa de la que depende el virus para sobrevivir. Para evitar este problema, el VPH codificados oncoproteínas, E6 y E7; juntas promueven la proliferación celular y previenen la apoptosis. La célula se vuelve permisiva para la replicación viral y se generan cientos o incluso miles de genomas de VPH dentro de una sola célula. Las proteínas de la cápside, L1 y L2, se expresan en las capas más superficiales del epitelio, donde tiene lugar el ensamblaje viral y finalmente se desprenden nuevas partículas virales infecciosas de la superficie epitelial22,60,61.
La expresión de las oncoproteínas E6 y E7 aumenta a medida que avanza el grado de lesión cervical desde neoplasia intraepitelial cervical de grado 1 (NIC-1) a NIC-3. Comúnmente, en las lesiones NIC-1 se conserva la capacidad de completar el ciclo de vida del VPH y de producir partículas virales infecciosas. El aumento de E6 y E7 en la infección por VPH-AR conduce a NIC-2 e impulsa la acumulación de cambios genéticos que contribuyen a la progresión del cáncer62.
Debido a la desregulación viral que se da en NIC-2/-3 se promueve la integración del ADN viral en el genoma de la célula hospedadora. En este proceso, el ADN viral sufre una ruptura de la doble cadena que principalmente se da sobre la región E1/E2 (63-65). E2 es el principal represor de la transcripción de E6 y E7; por lo tanto, en ausencia de este punto crítico de control se va a regular positivamente la transcripción de estos oncogenes61,66. (Figura 2)
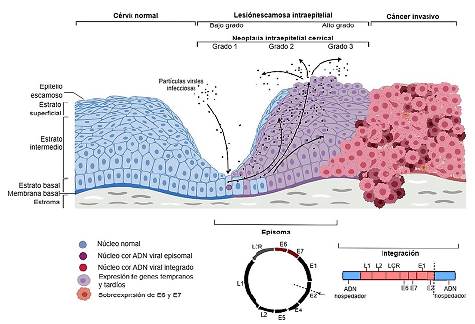
Se da una microabrasión para que entren los viriones de VPH en las células epiteliales mitóticamente activas de la capa basal. La infección es seguida por una fase inicial de amplificación del genoma, y luego por el mantenimiento del episoma viral en un número bajo de copias. La expresión de las proteínas E6 y E7 en las capas epiteliales superiores permite que la célula infectada vuelva a entrar en la fase S y aumente el número de copias del genoma viral. La finalización del ciclo de vida del VPH implica la expresión de las proteínas menor y mayor de la cápside (L2 y L1), la pérdida de control de la célula del ciclo celular y el empaquetamiento del genoma. En la enfermedad cervical, los niveles de expresión de E6 y E7 aumentan desde la neoplasia intraepitelial cervical de grado 1 a 3 (NIC1 a NIC-3). La desregulación viral en NIC2, y -3 facilita la integración del episoma en la célula hospedadora, lo que desregula aún más la expresión de los oncogenes E6 y E7. Adaptado de Sanjosé y colaboradores67.
Transformación maligna del queratinocito inducida por la infección crónica con VPH
La persistencia de la infección por VPH-AR es el factor de riesgo más importante para el desarrollo de lesiones premalignas que pueden evolucionar a cáncer68. Generalmente, menos del 10 % de las infecciones persisten durante 5 - 10 años69. Quienes mejoran la replicación y persistencia del VPH son las oncoproteínas E5, E6 y E7 cuya expresión induce la inmortalización y transformación maligna de los queratinocitos infectados del cuello uterino mediante la interacción con diferentes mecanismos celulares. Estas oncoproteínas carecen de actividad enzimática, pero actúan como proteínas adaptadoras que reclutan y secuestran las funciones de las proteínas del hospedador70-72.
E5 interactúa con algunos factores de proliferación celular, como el factor de crecimiento epidérmico y el factor de crecimiento derivado de plaquetas (EGF y PDGF, por sus siglas en inglés). Además, puede inhibir la apoptosis y la migración del complejo mayor de histocompatibilidad (CMH) a la superficie celular72,73.
Por otra parte, E6 interfiere con la función de numerosas proteínas celulares. La unión de E6 de VPH-AR a p53 promueve su degradación al reclutar el complejo enzimático necesario para la ubiquitinación y degradación proteosómica. En ausencia de este regulador negativo del ciclo celular, la proliferación celular no se interrumpe, lo que resulta en mutaciones y anomalías cromosómicas74,75. E6 también puede interactuar con quinasas, incrementando la tasa mitótica y puede promover la degradación de proteínas con dominios PDZ, involucradas en la regulación del crecimiento, la proliferación, la polaridad y la adhesión celular76. Adicionalmente, E6 induce un estado persistentemente activo de la telomerasa, contribuyendo a la inmortalización de los queratinocitos al mantener la longitud del telómero e incrementando el número de divisiones celulares77. E6 también se relaciona con el proceso de carcinogénesis por medio de diferentes oncogenes asociados a la transducción de señales celulares, entre los cuales se encuentra la familia RAS, Notch-1 y c-Myc78-80.
Finalmente, E7 codifica una oncoproteína potente que promueve la proliferación y supervivencia celular continua. Regula vías de señalización celular que median la degradación del supresor tumoral de retinoblastoma (pRB), provocando en los queratinocitos un estado de síntesis de ADN, atenuación de la respuesta de senescencia y evasión de los supresores tumorales70,81,82. Además, E7 también se une a c-Myc y media su actividad transcripcional, contribuye en la transición epitelio-mesénquima, induce la angiogénesis, desregula el metabolismo celular y promueve la amplificación de los centrosomas, dando lugar a mitosis multipolares e induciendo inestabilidad genómica y mutaciones, eventos que facilitan la progresión oncogénica66,83-87.
Evasión del sistema inmunológico por parte del VPH y modulación de vías metabólicas
VPH ha desarrollado mecanismos para evitar el reconocimiento inicial e interferir en la inmunidad del hospedador. Una de las formas en la que los genotipos de VPH-AR evitan la migración de células inmunitarias al sitio de la infección es regulando negativamente las citocinas proinflamatorias, incluido el Factor de Necrosis Tumoral α (TNFα) y positivamente las citocinas antiinflamatorias88.
Los principales mecanismos de evasión inmunitaria viral para la infección por el VPH están probablemente representados por la evasión del procesamiento y presentación de antígeno, el cambio de una respuesta inmunitaria Th1 a Th2, el silenciamiento de la respuesta inflamatoria a través del reclutamiento de células T reguladoras y la modulación de la apoptosis3. Sin embargo, uno de los mecanismos más importantes que demuestran el secuestro de la respuesta al ADN dañado y la desestabilización inmunitaria consiste en la desregulación de las respuestas al IFN89,90.
Como la mayoría de los virus de ADN, los VPH también han desarrollado estrategias efectivas para prevenir la síntesis de IFNs mediante la desregulación de la familia STAT que contiene importantes reguladores de la respuesta inmune innata91,92. Sobre la infección viral, E6 y E7 influyen directamente en la expresión del IFN-α e IFN-y porque son capaces de inhibir los factores de regulación del interferón 1 y 9 (IRF1, IRF9) e interfieren directamente con la vía JAK/STAT89. El oncogén E6 del VPH-18 funciona como un regulador negativo de la vía JAK/STAT activada por el IFN-a a través de la inhibición de la activación de Tyk2. También Tyk2 se asocia físicamente con E6. Esta interacción Tyk2/E6 es más fuerte en el VPH-18, que en VPH-11 y requiere de los dominios JH6-JH7 de Tyk2 para la unión al IFNAR1 (Figura 3) (93.
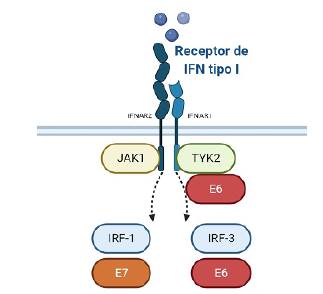
Figura 3 Interferencia de las proteínas de VPH en la respuesta al interferón. E6 se dirige a Tyk2, así como a IRF-3 y E7 se une a IRF-1.
Por otra parte, los cambios en las vías metabólicas celulares se adaptan a las necesidades específicas de cada virus, una vez se altera la vía metabólica se van a favorecer diferencialmente algunas etapas del ciclo replicativo del virus94. En este sentido, el VPH puede reconfigurar el metabolismo celular facilitando desde la entrada del virus a la célula, hasta la replicación y gemación viral95,96. Por ejemplo, se ha visto que al adicionar glucosa y glutamina a un cultivo de células HeLa infectadas es posible aumentar hasta en 40 000 veces la producción viral97. Actualmente, se sabe que los virus modulan el flujo de fuentes de carbono y promueven el cambio hacia la glutaminólisis, la síntesis de ácidos grasos y la glucólisis aeróbica, también conocida como efecto Warburg98, con el fin de producir energía, nucleótidos, aminoácidos y ácidos grasos necesarios para aumentar el ensamblaje, la propagación de partículas virales y el mantenimiento de infecciones latentes99. Por ejemplo, en la etapa transcripcional de genes tempranos se potenciará la glucólisis, mientras que, si necesita traducir estos genes, se promoverá la glutaminólisis y si lo que se requiere es producir viriones infecciosos, se activará la síntesis de ácidos grasos94.
Mecanismos de acción de los IFNs-I vs mecanismos de evasión de VPH
Interferón alfa
El IFN-α pertenece a la familia de IFN tipo I, es producido principalmente por leucocitos infectados por virus y participa en la síntesis del complejo mayor de histocompatibilidad clase 1 (MHC-1), encargado de la presentación antigénica durante la infección viral. En humanos existen 13 subtipos de IFN-α, compuestos por 189 aminoácidos y codificados por 14 genes no alélicos ubicados en el cromosoma 929,100.
Los IFN-α presentan una variedad de funciones biológicas directas o indirectas sobre diferentes células del sistema inmune incluidos los monocitos y las células asesinas naturales (NK), participando principalmente en la expresión antigénica mediada por MHC-1. Debido a su actividad inmunomoduladora, en la última década este interferón ha sido considerado como una prometedora opción terapéutica para el control de enfermedades virales y tumorales, presentando mayor efectividad respecto a los demás tipos de interferón100. La vida media de este interferón es corta, por lo que se requieren dosis frecuentes para el control de enfermedades. Sin embargo, se han realizado modificaciones para aumentar su vida media en circulación o prolongar el tiempo de liberación101.
Los IFN-α están presentes en las infecciones causadas por VPH. Se ha demostrado que las células dendríticas de tejidos con CCU infectadas con partículas similares al virus VPH-16 (VLP), secretan varias citocinas incluyendo IFN-α, IL-6, TNF-α e IL-8 después de la activación de la señalización dependiente de MyD88 (Figura 4) (102-104.
Adicionalmente, Sen y colaboradores han comprobado su efecto anti-VPH al evaluar al IFN alfa-n3 y alfa-2b sobre cultivos tridimensionales infectados con VPH-16, -18 y -31. Se reportó un efecto inhibitorio sobre la replicación viral desde el 50 hasta el 80 %. Este porcentaje se asoció directamente a las concentraciones de IFN implementadas y al genotipo de VPH a tratar105.
No obstante, VPH también ha desarrollado mecanismos para protegerse de la acción de los IFN-α. Se ha encontrado que E6 de VPH-18 inhibe la vía Jak-STAT inducida por IFN-α, debido a su capacidad para interactuar e impedir la activación de Tyk2 (Figura 1) (93. E6 de VPH-18 puede disminuir la transactivación de diferentes ISGs mediados por IFN-a incluidos el ISG-15 y el gen de la proteína 53 inducida por IFN (IFP-53) (93.
Igualmente, los VPH-AR al mejorar la expresión del regulador del desarrollo relacionado con IFN 1 (IFRD1), disminuyen la acetilación de K310 del NF-kB en los queratinocitos, reduciendo su translocación nuclear y resultando finalmente en una regulación negativa de los IFN-α y de otras citocinas proinflamatorias106. También se ha establecido que los dominios CR1 y CR3 de E7 juegan un papel en el deterioro de la acetilación de k310. Al inducir la transactivación del NF-kB con imiquimod, esta fue inhibida por E7, disminuyendo consecuentemente los niveles de IFN-α, IL-6 y TNF-α (Figura 4) (107.
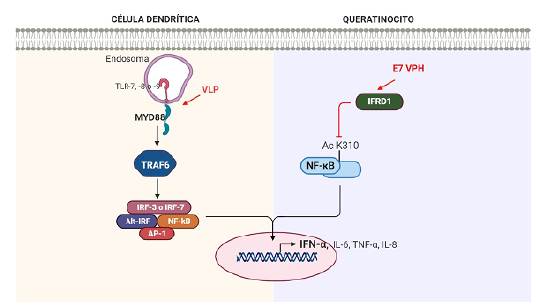
Figura 4 Regulación positiva de IFN-α en células dendríticas de tejidos con CCU infectadas con partículas similares al virus VPH-16 (VLP) (izquierda). Regulación negativa de los IFN-αy de otras citocinas proinflamatorias en los queratinocitos de cuello uterino infectados.
Por otra parte, mediante análisis de microarreglos y análisis de transferencia Northernblot en diferentes líneas celulares infectadas con VPH-31, se descubrió una regulación negativa en el nivel basal de expresión de varios ISGs, incluidos los genes que codifican para MxA, la proteína 27 inducible por IFN-α (IFI-27), la proteína inducible por interferón 56 (P56), OAS-2, STAT-1, entre otros (Figura 1) (108. Dado que MxA es inducido principalmente por IFN-a, se trataron las células con este IFN109. Se encontró que los niveles de MxA y STAT-1 se veían afectados en respuesta al tratamiento en las células positivas para VPH-31. A dosis más altas de IFN-a durante un período prolongado, la vía de transducción de señales de IFN pudo activarse en células positivas para VPH-31108.
Conociendo las vías por las que VPH puede modificar la respuesta celular mediada por IFN-α, facilitar su replicación y evadir la detección por parte del sistema inmune, es posible generar nuevos blancos terapéuticos y optimizar las estrategias terapéuticas actuales contra CCU.
Interferón beta
Se ha demostrado que tanto IFN-α como IFN-β inducen apoptosis en células sensibles y se cree que esto ocurre de manera específica para cada tipo de célula110,111. Además, se ha demostrado que IFN-β tiene una mayor eficacia en el control de lesiones derivadas del VPH en ensayos clínicos que IFN-α112,113.
Al analizar los efectos de IFN-p en las células positivas para VPH-31 se encontró que el tratamiento con IFN-β dificulta la entrada de VPH y promueve la eliminación de los episomas de VPH latentes en células infectadas de manera persistente114. En otro estudio con queratinocitos cervicales infectados con episomas de VPH-16, se evidenció que el tratamiento con IFN-β conduce a una rápida reducción en el número de episomas y a la aparición de integrantes latentes115.
Esto sugiere la posibilidad de que el tratamiento con IFN-p in vivo pueda seleccionar células que mantengan copias integradas del ADN del VPH. Y debido a que se ha propuesto como una terapia para las lesiones derivadas del VPH, debe manejarse con precaución para evitar la resistencia tras un tratamiento prolongado114,116.
Por otro lado, se ha visto que varios miembros de la familia APOBEC3 (A3) citidina desaminasa inducible por IFN-β editan el genoma de VPH en queratinocitos cervicales y en lesiones cervicales VPH positivas117.
Particularmente A3A, uno de los miembros de esta familia, restringe significativamente la infección por VPH118.
Interferón épsilon
La estructura del IFN-ε consta de 192 aminoácidos y solo tiene un 30 % de aminoácidos concordantes con una secuencia consenso de IFN-α y -β. Sin embargo, se clasificó en la familia de IFN tipo I debido a la localización del gen y porque su señalización se da través de IFNAR1 e IFNAR28,119,120.
A diferencia de otros IFN tipo I, el IFN-ε no es inducido por vías conocidas de RRP, sino que se expresa constitutivamente en los pulmones, el cerebro, el intestino delgado y los tejidos reproductivos, y es regulado hormonalmente8,120. También se han encontrado grandes diferencias en los perfiles de expresión génica entre los cultivos de células humanas tratadas con IFN-ε y los tratados con IFN-α-2b. Además, la regulación diferencial de los genes relacionados con el sistema nervioso central por parte del IFN-ε humano recombinante (rhIFN-ε) sugiere un papel del IFN-ε en el mantenimiento de la estructura y función cerebral121.
En un estudio sobre las actividades biológicas del rhIFN-ε se analizaron las actividades antivirales, antiproliferativas y de activación en células NK. Se determinó que rhIFN-ε tenía una actividad antiviral específica de aproximadamente 6x105 UI/mg en el sistema WISH/VSV. En cuanto a las actividades antiproliferativas y de promoción de la citotoxicidad de las células NK in vitro, fueron menos potentes al compararse con rhIFN-α. También se analizaron los patrones de expresión génica en células tratadas con ambos IFNs, se encontraron 22 278 transcriptos humanos modulados en las células tratadas con rhTFN-ε, lo cual fue inferior a lo detectado en las células tratadas con rhIFN-α121,122.
Se demostró que IFN-e es el único miembro de la familia de IFN tipo I expresado constitutivamente en las células HeLa en bajos niveles y que su expresión está regulada al alta después de la estimulación con TNF-α. Se encontró que al silenciar la expresión de IFN-ε disminuye la fosforilación y expresión de STAT1 y se inhibe la expresión de RIG-I. Proponiendo un mecanismo secuencial en el que el ARNm de IFN-ε es estabilizado por TNF-a, aumentando la síntesis de IFN-ε, el cual se une al receptor de IFN tipo I, aumentando STAT1 y finalmente regulando la expresión de RIG-I123.
Para evaluar el papel de IFN-ε en la inmunidad de las mucosas se infectaron ratones vía intranasal con el virus Vaccinia coexpresando IFN-ε. Se observó un rápido aclaramiento del virus que estuvo relacionado con la inducción de una población CD8+CD107a+IFN-y+ específica, un gran reclutamiento de linfocitos y un subconjunto intensificado de células T CD8+ y CD4+. Debido a que inicialmente se encontró IFN-e en el intestino y tejidos reproductivos, se midió la presencia de células T CD8+ específicas de antígeno en las placas de Peyer, y se encontró que el IFN-ε fue el responsable de su migración al intestino. Este estudio permite establecer el papel fundamental de IFN-ε en la mucosa y su posible uso en el control de infecciones pulmonares, intestinales y en enfermedades de transmisión sexual124. Además, se ha encontrado que el líquido seminal regula diferencialmente la expresión del IFN-ε en células epiteliales cérvico-vaginales humanas125. Por tanto, es de esperarse que el IFN-ε tenga un papel protector en el tejido reproductivo.
Fung y colaboradores quisieron determinar si el IFN-ε era inducido por RRP, como la mayoría de IFN tipo I Sin embargo, tras la estimulación con activadores de RRP, no hubo cambios en la expresión de IFN-ε, tampoco con factores de transcripción de la familia IRF, ni tras la infección con Chlamydia muridarum, el virus del bosque Semliki o VHS-2. De manera consistente encontraron que el promotor proximal de IFN-ε carecía de elementos de respuesta para estas vías; por lo tanto, quisieron comprobar su expresión constitutiva, la cual fue detectada en el útero, el cuello uterino, la vagina y el ovario. Además, la inmunohistoquímica del endometrio demostró la expresión de IFN-ε en las células epiteliales luminales y glandulares8.
Posteriormente, al administrar estrógenos a ratones hembra ovariectomizadas, notaron que la expresión de IFN-ε aumentó 6 veces, sugiriendo que tiene regulación hormonal. Para comprobar esto en humanos, se analizaron células epiteliales del endometrio de mujeres en diferentes etapas del ciclo menstrual o posmenopáusicas, encontrándose una mayor expresión de IFN-ε en la fase proliferativa, cuando la concentración de estrógenos es mayor, y niveles indetectables en mujeres posmenopáusicas. De igual manera, IFN-e se expresó en líneas celulares derivadas de cáncer de endometrio8.
En este estudio también se determinó el papel de IFN-ε en infecciones virales (VHS-2) y bacterianas (Chlamydia), encontrándose un rol protector en ambos casos: el virus solo fue detectable en ratones sin IFN-e y no en el tipo silvestre; igualmente, la detección de bacterias en los frotis vaginales fue mayor en ratones sin IFN-ε, mientras que el número de células NK, cuyo papel protector es conocido en esta infección, disminuyó. En este caso, en los ratones de tipo silvestre la expresión de ARN de IFN-ε no cambió, lo que concuerda con el hecho de que IFN-e no está regulado por las vías RRP8.
Para confirmar estos hallazgos en humanos, se tomaron muestras cérvico-vaginales de mujeres embarazadas y se midieron las concentraciones de IFN-ε, tanto en mujeres sanas como en mujeres con VHS. Se encontró un aumento de IFN-ε durante el embarazo, que fue mayor en las mujeres sanas en comparación con aquellas que tenían VHS126.
La relación entre una mayor susceptibilidad a las infecciones del sistema reproductor femenino (SRF) en mujeres que consumen anticonceptivos con progestágenos es conocida127. Esta relación podría explicarse por una disminución en los niveles de INF-ε, como se ha visto en los modelos de infección del SRF pretratados con progesterona8.
En un estudio sobre el efecto de rhIFN-ε en la infección por VIH, se evidenció que este afectó diferentes etapas del ciclo de replicación del virus después de la entrada a la célula hasta la traducción a proteínas virales, e incluso disminuyó la infectividad de los viriones. De acuerdo con los resultados, la razón fue el aumento de factores de restricción contra VIH causado por INF-ε, cuya potencia de inducción fue parecida a la de IFN-α y -β128.
Como se ha demostrado, las características únicas de IFN-ε le otorgan propiedades protectoras contra infecciones de transmisión sexual (ITS) como Chlamydia muridarum o Virus Herpes simplex tipo 2 (VHS-2) (8. El papel de estas ITS como posibles cofactores en el proceso de carcinogénesis cervical inducido por la infección con VPH129, sumado a la expresión constitutiva de IFN-ε en las células epiteliales de la mucosa genital, sitio en el que ocurre el primer contacto con agentes infecciosos, aumenta la eficacia de IFN-ε en el control de estos patógenos, y podría facilitar la creación de estrategias preventivas o de tratamiento contra el cáncer de cuello uterino (CCU) (8,123.
Interferón kappa
El IFN-k hace parte de la familia de los IFN tipo I, comparte con estos una homología del 30 % y su gen se encuentra adyacente en el cromosoma 9 traduciendo una citoquina de 180 aminoácidos que se expresa de manera constitutiva y predominante en queratinocitos, a diferencia de otros IFNs como el IFN beta (IFN-β) que se induce en respuesta a estimulación y está presente en diferentes líneas celulares5,130.
Muchas de las vías reguladas positivamente por el IFN-k a través de la estimulación de factores de transcripción como STAT1 e IRF1 son importantes en la respuesta antiviral. Gracias a estudios de transcrip-toma se conoce que el 71 % de los 1 367 genes estimulados por IFN-k coincide con los genes regulados en general por los IFN tipo I. Esto sitúa evidentemente al IFN-k dentro de la familia de los IFN tipo I cuyas funciones antivirales se dan principalmente en los queratinocitos131. Sin embargo, también en monocitos y células dendríticas puede estimular la producción y liberación de factor de necrosis tumoral alfa (TNF-α) y de interleucina-10 (IL-10) e inhibir la IL-12. Debido a su fuerte unión a la heparina, se sugiere que el IFN-k se retiene en el sitio de producción y que activa las vías de señalización por contacto intercelular, estimulando respuestas antivirales localizadas en el sitio de la infección132.
Además, por el papel modulador del IFN-k en la respuesta inflamatoria también se han asociado mutaciones en este gen a enfermedades autoinmunes como lupus eritematoso133 y artritis reumatoide134.
Asimismo, por inmunohistoquímica se ha encontrado que es muy débil o indetectable en la piel sana, en la psoriasis y en la dermatitis atópica, pero está expresado fuertemente en los queratinocitos basales y suprabasales en la dermatitis de contacto alérgica y en el liquen plano135.
Actualmente, se ha establecido una asociación entre las lesiones premalignas y cancerosas inducidas por VPH y la supresión del IFN-k. Por medio de análisis del transcriptoma de células con VPH-AR se encontró una menor expresión de los ISGs causada por la disminución del IFN-k. También se ha determinado que el IFN-k puede ser inhibido por VPH-16 y -18 por medio de la proteína E2, encargada de regular la transcripción y replicación del virus136.
Se identificó por primera vez la regulación del IFN-k en el contexto de la carcinogénesis inducida por VPH utilizando cultivos primarios de queratinocitos del prepucio humano (QPH) inmortalizados por la expresión de las oncoproteínas E6 y E7, de manera individual o sumada, y células de carcinoma de cérvix13. En presencia del oncogén E6 y en las líneas celulares de carcinoma de cérvix, el IFN-k estuvo regulado negativamente, contrario a lo encontrado en presencia del oncogén E7 en donde no hubo cambios en su expresión. Además, se evidenció mediante secuenciación por bisulfito que en las células que codifican el oncogén E6 de VPH-16 se induce la metilación de novo en una región que contiene islas de CpGs, adyacentes al sitio del inicio de la transcripción, inhibiendo así epigenéticamente la expresión de IFN-k. Este tipo de inhibición pudo revertirse después de demetilar el ADN de las células positivas para E6, E6/E7 y líneas celulares de carcinoma cervical usando inhibidores de la enzima ADN metil-transferasa-1 (DMNT-1) o inhibiendo E6 usando ARN de interferencia, sugiriendo la participación directa de la oncoproteína E6 en el silenciamiento epigenético de IFN-k (Figura 5) (13.
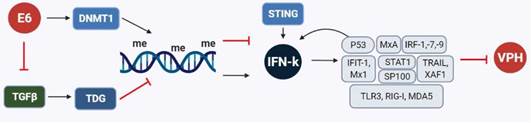
Figura 5 Silenciamiento epigenético de IFN-k y mecanismos regulatorios en la transcripción y expresión de proteínas claves en la respuesta antiviral.
En contraposición, la re-expresión de IFN-k en células de carcinoma cervical fue relevante a nivel funcional ya que reconstituyó la respuesta antiviral, aumentando el nivel de proteína p53, MxA y de los factores reguladores de INF-1, -7 y -9. Esto se revirtió en células positivas para E7, se suprimió la expresión constitutiva tanto del IFN-k como de p53 utilizando ARN interferentes, encontrándose el efecto inverso en los niveles de estas proteínas indispensables en la respuesta inmediata y tardía mediada por IFN. Todo esto sugiere la existencia de un circuito de retroalimentación positiva entre IFN-k, p53 y componentes de la vía de señalización del IFN-k para mantener un estado basal antiviral (Figura 5). Al estimar la relevancia clínica de los datos in vitro en biopsias de pacientes con CCU en comparación con biopsias de tejido normal, se corroboró una relación inversa entre la expresión del IFN-k y la transcripción de VPH13, revelando una estrategia novedosa sobre cómo los VPH-AR pueden eliminar la respuesta inmune innata en sus células hospederas.
Adicionalmente, se pudo conocer por análisis de microarreglos de ARN que VPH inhibe a nivel transcripcional tanto al IFN como al TGFβ A partir de esto se descubrió la relación entre estos dos sistemas y cómo la vía de señalización del TGFβ induce la desmetilación del promotor del IFN-k por medio de la timina ADN glicosilasa (TDG). De esta manera la transcripción y expresión del IFN-k logra aumentarse cuando el TGFβ. se está expresando, provocando una supresión de la transcripción viral (Figura 5) (137.
También se ha determinado la expresión del IFN-k en el espectro completo de la carcinogénesis cervical, incluyendo tejido normal, displásico y carcinoma ex vivo de biopsias cervicales completas, estroma y epitelio cervical microdiseccionado138. Aquí se encontró una mayor prevalencia y niveles de expresión del ARN mensajero de IFN-k en las biopsias de tejidos con algún grado de enfermedad en comparación con tejidos sanos VPH negativos. Sin embargo, en la microdisección por captura láser se observó que el IFN-k inducido por VPH se expresó a nivel estromal mientras que en el epitelio enfermo el IFN-k estuvo ausente, demostrando que los responsables de la expresión del IFN-k en el estroma afectado son los monocitos y las células dendríticas. En definitiva, en las células de carcinoma cervical no se observó un cambio en la expresión de IFN-k, representando un hallazgo importante que favorece la progresión de la enfermedad138.
Los análisis de transcriptomas en queratinocitos con VPH-16, -18 y -31 indican una modulación negativa de diferentes ISG, no solo de aquellos con función directa antiviral o implicados en la señalización del IFN y apoptosis, sino también sobre los que inducen los RRP, que a su vez pueden inducir la expresión de IFN-β y -λ en los queratinocitos (respectivamente: IFIT1 y Mx1; STAT1; TRAIL y XAF1; TLR3, RIG-I y MDA5) (Figura 5). Esta regulación negativa se asoció principalmente a la acción del oncogén E6, el cual inhibió la expresión constitutiva del IFN-k, que al re-expresarse en células VPH positivas resultó en la reactivación de los ISG6.
Con el fin de comprender más acerca del mecanismo inhibitorio de E6 sobre el IFN-k se compararon células de carcinoma cervical con células obtenidas de una neoplasia intraepitelial cervical grado 1 (NIC-1). Al activar el oncogén E6, se obtienen 1 000 veces menos transcripción de INF-k en células cancerosas, en las que posteriormente al usar 5-Aza-dC solo se pudo activar 9 veces la transcripción. Estos resultados comprueban la importancia del papel de la metilación del ADN en la inhibición de la transcripción del IFN-k. Sin embargo, otros estudios sugieren la posibilidad de otros mecanismos afectados que no permiten un aumento significativo en la transcripción aún en ausencia de metilación del promotor de IFN-k6. Además, se demostró que la disminución de la expresión del IFN-k no solo se da en fenotipos malignos, ya que en células no malignas con replicación persistente de VPH la expresión está disminuida6.
Complementariamente, se ha sugerido que los VPH-AR tienen como blanco al IFN-k para evitar la expresión de proteínas como Sp100 o como IFIT-1, la cual tiene efectos inhibitorios sobre E1 de VPH-16, -18 y -31, y puede afectar la oligomerización de E1, la unión al ADN o la propia actividad helicasa, provocando finalmente la inhibición de la replicación del ADN de VPH131,139,140.
Al evaluar el perfil de expresión génica de queratinocitos VPH positivos se ha demostrado que el dominio de trans-activación amino-terminal de la proteína E2 de VPH-16 y -18 disminuye la expresión de STING y de IFN-k y, en consecuencia, se inhibe la transcripción de los ISG. Además, este estudio reveló que al silenciar STING se regulaba la baja la transcripción del IFN-k en un 20 %, sugiriendo a STING como un regulador positivo del IFN-k. Igualmente, al evaluar estos hallazgos in vivo, se encontró una supresión de la expresión del IFN-k en tejidos normales VPH positivos y en lesiones escamosas intraepiteliales de bajo grado en comparación con tejidos VPH negativos136. Nuevamente, se sugiere que el silenciamiento del IFN-k es crucial en la progresión de lesiones por VPH y que en este suceso están implicadas las proteínas tempranas de VPH.
Conclusiones
En los últimos años, se ha descubierto que el VPH ha generado mecanismos complejos de patogenicidad a través de los cuales causa un impacto negativo en la respuesta inmune del hospedero, uno de estos es contrarrestar los IFN tipo I.
Se ha estudiado la interacción de los IFNs-α,-β,-ε,-K frente al VPH y se ha encontrado que estos cumplen funciones específicas necesarias para la inmunidad del huésped: el IFN-α es un inductor importante de ISGs y tiene efectos inhibitorios sobre la replicación de VPH; el IFN-β puede generar una fuerte reducción en el número de episomas de VPH; el IFN-e tiene actividad en las mucosas y aumenta la expresión de células T citotóxicas, actuando indirectamente en la inmunidad adaptativa frente al VPH; y el IFN-K, se expresa constitutivamente en los queratinocitos, inhibe los mecanismos de patogenicidad del VPH y restaura la actividad antiviral, disminuyendo la proliferación celular, la transcripción y las copias del genoma viral.
Es importante desentrañar las bases moleculares de las funciones de los IFN tipo I en la patogénesis mediada por VPH, ya que proporciona información valiosa sobre posibles blancos moleculares que a futuro podrían ser útiles en la creación de terapias más efectivas que puedan bloquear procesos metabólicos específicos y claves para la infección y propagación de VPH, sin que los efectos secundarios de estos nuevos protocolos afecten profundamente el funcionamiento y supervivencia del paciente infectado con VPH.

