











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
En las últimas décadas la obesidad es considerada una enfermedad crónica que presenta una elevada prevalencia a nivel mundial, por esta razón es una de las principales amenazas para la salud en la mayoría de las sociedades alrededor del mundo. Es conocida como la epidemia del siglo XXI, adicionalmente, esta enfermedad produce grandes costos para los sistemas de salud1.
El desarrollo de la obesidad se ha relacionado con la expansión del tejido adiposo que conlleva a la elevación en la producción de citoquinas proinflamatorias, que junto a los ácidos grasos son los responsables del desarrollo de la resistencia a la insulina y por tanto DMT22,3. El proceso que favorece el desarrollo de obesidad a nivel fisiopatológico, inicia con la respuesta inflamatoria en los adipocitos y posteriormente se activa un reclutamiento de macrófagos, esto da como resultado un estado inflamatorio crónico de bajo grado3. El papel inflamatorio de los adipocitos está relacionado con su expansión, hiperplasia (incremento en número) e hipertrofia (incremento en tamaño-volumen). Estos factores modifican la capacidad de secreción de adipocinas en los adipocitos, que actúan localmente (autocrino / paracrino) y también sistémicamente (endocrino)4,5. Un mecanismo importante del estado proinflamatorio consiste en la activación de las vías inflamatorias que resulta en estrés del retículo endoplásmico (RE) y estrés mitocondrial, debido a la demanda en el retículo endoplásmico adipocitario y a la sobrecarga de la capacidad funcional5,6. En condiciones donde hay sobrecarga de grasa, los adipocitos secretan citocinas pro-inflamatorias adicionales y ácidos grasos libres, también expresan nuevos receptores para diversas señales endocrinas y neuronales, donde se altera la estructura, función y regulación de factores de transcripción. De esta manera se alteran las señalizaciones moleculares desarrollando la patogénesis de la obesidad y la DMT26,7
Un mecanismo importante al inicio de la inflamación en la obesidad es el estrés oxidativo. El incremento en la captura de glucosa por los adipocitos en condiciones de hiperglucemia causa un exceso de especies reactivas de oxígeno en las mitocondrias, lo cual condiciona daño oxidativo y activa señales de inflamación dentro del adipocito6. El daño en el adipocito causa quimiotaxis de macrófagos y exacerba aún más la inflamación local. La hiperglucemia también estimula la generación de especies reactivas de oxígeno en los adipocitos, con lo cual se incrementa la producción de citocinas proinflamatorias7.
En modelos animales el retículo endoplásmico conduce a la activación de la cinasa de c-jun NH2-terminal (JNK) y la degradación de la proteína inhibidora de la cinasa del NF-kB (IkB) o IKK, esto genera la activación del factor nuclear kappa B (NF-kB). Este último, es un factor de transcripción inducible que participa en la respuesta inmune y en la respuesta inflamatoria, ya que activa un gran número de citocinas proinflamatorias y suprime otras, como adiponectina y, también, los transportadores de glucosa tipo 4 (GLUT-4). Esto ha sugerido que el proceso inflamatorio está estrechamente asociado a la obesidad y contribuye a la resistencia a la insulina8. Así mismo, la insulina participa en la homeostasis de la glucosa, mediante el estímulo del transporte de glucosa y el consumo de energía por medio de PI3 K/AKT, la activación de la vía AKT es un mediador central de actividades metabólicas y de la insulina, estimula la translocación de GLUT4 a la membrana plasmática, por lo tanto, AKT aumenta la entrada de glucosa en la célula8.
Aunque la resistencia a la insulina es característica de la obesidad y la DMT2, es fundamental identificar las vías de señalización y los factores de transcripción que podrían permitir tales acciones alteradas de la insulina. Además, el estudio fisiopatológico del tejido adiposo en muestras directas de pacientes se dificulta en gran medida, por tal razón existen diferentes modelos experimentales in vivo e in vitro (TNF-alfa, Tapzigargina, oleato, palmitato, H2O2, entre otros) que buscan simular las condiciones patológicas de obesidad y diabetes9-11. Sin embargo, es necesario implementar modelos in vitro que logren imitar, al menos en parte, los estados fisiopatológicos de las células adipocitarias para poder realizar aproximaciones terapéuticas futuras. El objetivo de este estudio fue realizar y caracterizar un modelo in vitro de obesidad e insulino resistencia empleando altas concentraciones de glucosa e insulina para simular el estado de obesidad e insulino resistencia, y generar un modelo sencillo, práctico, económico y disponible para la futura evaluación de moléculas con potencial farmacológico, así como base para el estudio de los mecanismos moleculares implicados en la insulino-resistencia.
Métodos
Cultivo celular
Los adipocitos 3T3-L1 de tipo fibroblasto procedentes de embriones de ratón obtenidos de la American Type Culture Collection, ATCC CRL-3242, se sembraron a una densidad de 3000 células/cm2 en medio DMEM [Dulbecco's Modified Eagle's Medium (SIGMA)], enriquecido con bicarbonato sódico a 1,5 mg/Ml, L-glutamina al 2 % (m/v), 10 % de suero bovino fetal (SFB) (v/v) y antibiótico-antimicótico al 1 °% (v/v). Todos los cultivos permanecieron en incubación a 37 °C, con 5 % de CO2 por 3 días, hasta obtener 80 % de confluencia, luego se estimuló su diferenciación12. Según protocolo de diferenciación establecido por Moreno Castellanos N.12; las células se incubaron por 3 días en el medio DMEM descrito anteriormente, con 3-isobutil-1-metilxantinan 0,6 mmol / l (IBMX), Insulina 1 μg /ml (m/v), Dexametasona 0,2 μmol / l (m/v) y Rosiglitazona 2 μmol/1 (m/v), luego del primer estímulo las células fueron alimentadas cada 3 días con el mismo medio, pero sin IBMX, dexametasona y rosiglitazona, hasta completar 10 días restantes de diferenciación para iniciar con todos los experimentos. El diseño experimental fue desarrollado en la Universidad Industrial de Santander y aprobado por el Comité de ética de esta institución (4110/20 de septiembre 2019).
Modelo de inducción de alta glucosa - alta insulina (HGHI)
Para inducir el modelo de obesidad en los adipocitos diferenciados se incubaron por 24 horas a 37 °C, con 5 °% de CO2 con tratamiento en el medio de cultivo DMEM empleando concentraciones basales de glucosa (100 mg/dL) e insulina (104 pmol/L) como control (sin tratamiento HGHI) y concentraciones altas de glucosa (450 mg/dL) e insulina (106 pmol/L) como tratamiento alta glucosa - alta insulina (tratamiento HGHI). Después de este tiempo de incubación se iniciaron ensayos tanto de las células tratadas con el modelo de inducción de alta glucosa - alta insulina como los controles (sin tratamiento HGHI).
Ensayo de viabilidad celular (actividad metabólica) por MTT
El efecto del tratamiento de HGHI sobre la viabilidad celular de los adipocitos se evaluó mediante la técnica de MTT [Bromuro de 3-(4,5- dimetiltiazol-2-ilo)-2,5-difeniltetrazol], como se ha descrito previamente13. Una vez las células llegaron a confluencia, se indujo la diferenciación a adipocitos maduros y posteriormente se realizó el tratamiento con el modelo HGHI, para esto los adipocitos fueron tratados o no durante 24 horas con el modelo de HGHI, luego incubados con solución de 0,5 g/L (m/v) de MTT, por cinco horas a 37 °C, tras este tiempo, las células fueron lavadas y posteriormente se aplicó DMSO (Dimetil Sulfóxido) y finalmente se realizó la lectura de absorbancia a 570 nm.
Tamaño celular
Los adipocitos maduros con o sin medio de inducción HGHI se tiñeron con Oil Red O para medir el diámetro mediante microscopía óptica por medio de métodos de densitometría. Brevemente las células se fijaron en paraformaldehído al 4 % (m/v) a temperatura ambiente en solución salina tamponada con fosfato [PBS 0,01 mmol/l, pH 7,4] durante 15 minutos, se lavaron con isopropanol (Sigma) al 60 % (v/v) y se tiñeron con Oil Red O (Sigma) durante 30 minutos. Las células se visualizaron con el microscopio óptico Leica DM500 (Leica), para finalmente ser analizadas por densitometría con el software Image J.
Movilización de lípidos
Para evaluar la movilización de lípidos, se realizó evaluación en adipocitos maduros de la lipogénesis (contenido de triglicéridos) y la lipólisis (liberación de glicerol), éstas dos técnicas fueron evaluadas en células tratadas y no tratadas con HGHI. Se llevó a cabo el protocolo descrito previamente13. En concreto, a los adipocitos maduros tratados con HGHI se les retiró el medio de cultivo y se almacenó para la posterior cuantificación de la lipólisis. Para la lipogénesis, algunas células fueron tratadas (modelo HGHI) y otras no tratadas con HIGH (modelo basal), posteriormente los dos modelos fueron fijados con PFA al 4 % (m/v), a continuación, se añadió el isopropanol al 60 % (v/v) seguido de Oil Red O (Sigma) (v/v) durante 30 minutos, para revelar con isopropanol al 100 % (v/v) y finalmente leer las muestras a 540 nm.
Por otro lado, se evaluó la actividad lipolítica en los diferentes grupos experimentales en el medio de cultivo obtenido de las mismas células en las que se evaluaron los triglicéridos. Esta técnica se basa en la cuantificación del glicerol libre mediante un método colorimétrico, para lo cual se utilizó el kit Free Glycerol Reagent (Sigma) como se ha descrito previamente13. Brevemente se añadió el reactivo de glicerol, y se dejó incubar durante 15 minutos a temperatura ambiente, finalmente se realizó lectura a 540 nm mediante espectrofotometría.
Western blotting
Los adipocitos tratados y no tratados con HGHI, se disolvieron y homogeneizaron empleando Tritón (20 mm de Tris, pH 7,4, 150 mm de NaCl, 1 % de Tritón e inhibidor completo de proteasas), para luego ser incubados con DNasa I (Sigma) durante 15 minutos. Posteriormente las proteínas fueron cuantificadas empleando el método Bradford. Por medio electroforesis en gel de dodecil sulfato de sodio-poliacrilamida al 10 % (SDS-PAGE) en condiciones de desnaturalización se separaron 25 μg de proteínas para luego ser bloqueadas a temperatura ambiente por 1 hora, con solución salina de Tris con 0,05 % de Tween-20 (TBS-T) con un 5 % de leche en polvo. Los blots obtenidos se incubaron durante la noche a temperatura de 4 °C con anticuerpos primarios de las proteínas a evaluar (Akt,Cell Signaling), fosfo (Thr308)-Akt (pAkt, Cell Signaling), c-Jun N-terminal quinasa (JNK, RD Systems) fosfo (Thr183 y Thr185)-JNK (pJNK, Santa Cruz), sustrato del receptor de insulina 1 (IRS-1, Santa Cruz), fosfo (Tyr 612) IRS-1 (pIR, Cell signalling), proteína homóloga de C/EBP (CHOP, Santa Cruz), glutatión sintetasa (GSS, Abcam) y P-Actina (Sigma), para luego retirar el anticuerpo primario y posteriormente incubar la membrana con el anticuerpo secundario. Finalmente, los complejos antígeno-anticuerpo se observaron por medio de anticuerpos anti-conejo, anti-ratón o anti-cabra IgG conjugados con peroxidasa de caballo (Abcam) según correspondiese, el kit de quimioluminiscencia mejorada (ECL-Pls, Ge Healthcare) y películas de autorradiografía (Sigma) permitieron visualizar las bandas. Por medio de un análisis densitométrico con el software Gel DocTM gel documentation system-Quantity One 4.5.0 (Bio-Rad) se determinó la intensidad relativa de las bandas y se normalizó con los valores de densidad de P-actina.
Análisis estadístico
Los datos de este trabajo se expresan como media ± SEM (error estándar de la media), el análisis estadístico se realizó empleando GraphPad Prism versión 7 y SPSS, para determinar la diferencia estadística entre las diferentes variables numéricas se empleó ANOVA bidireccional o unidireccional, seguido por las pruebas de Scheffé o Dunnett, si la distribución de los datos no era normal se empleó Kruskal-Wallis seguida de comparaciones por pares de Mann-Whitney U. Los valores se consideraron estadísticamente significativos cuando p <0,05.
Resultados
Ensayo de viabilidad celular
Para evaluar el efecto del tratamiento de HGHI sobre la viabilidad de los adipocitos, se determinó el ensayo MTT [bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio] que se basa en la capacidad de las células metabólicamente activas para reducir el colorante soluble en agua [bromuro de 3- (4,5-dimetiltiazol-2-il) -2,5-difeniltetrazolio] formando cristales de formazán violeta no solubles en agua dentro de la célula, se siguió el protocolo descrito previamente14. Los datos representados en la Figura 1 corresponden al porcentaje de viabilidad celular del modelo control respecto al modelo HGHI (Figura 1a). Se observó un incremento en la viabilidad celular y en porcentajes de viabilidad más altos en las células tratadas con HGHI hasta llegar a 338,96 % en comparación con el control. Por lo tanto, se obtuvo un aumento estadísticamente significativo en la viabilidad de los adipocitos tras inducción con el modelo de HGHI respecto al modelo basal, demostrando una tolerancia óptima al tratamiento y modelo in vitro establecido.
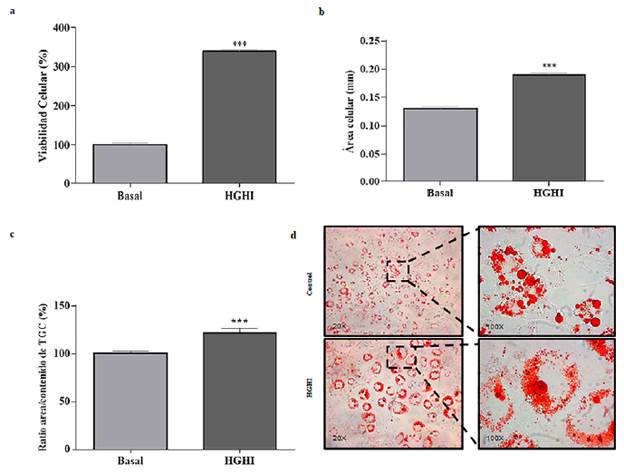
Figura 1 Caracterización del modelo HGHI in vitro. (a) viabilidad celular, (b) diámetro celular, (c) Ratio de área celular/ contenido de triglicéridos (TGC) y (d) morfología de las células con coloración de triglicéridos, Oil Red-O. Los valores evaluados representan la media ± SEM. Las diferencias entre grupos se analizaron mediante ANOVA de una vía seguido de la prueba de Dunnett: *P<0,05; **P<0,01; y ***P<0,001 respecto al control.
Tamaño celular
Con el fin de estimar la hipertrofia se evaluó el diámetro celular como se puede observar en la (Figura 1b), donde se evidencia un aumento de diámetro en los adipocitos tratados con HGHI en promedio de 0,19 mm (DS 0,009) en comparación a los adipocitos control con un diámetro promedio de 0,13 mm (DS 0,011), se demostró un aumento estadísticamente significativo del 46 % en los adipocitos tratados con HGHI respecto al control. Adicionalmente, las imágenes teñidas con Oil Red O que se muestran en la (Figura 1d), son evidencia de la hipertrofia de los adipocitos después del tratamiento con HGHI, en comparación a los adipocitos normales, debido a el tamaño aumentado y un mayor número de gotas lipídicas (aumento en la acumulación de triglicéridos) que se pueden observar en los adipocitos tratados con HGHI, (Figura 1c).
Movilización de lípidos
Para evaluar la movilización de ácidos grasos de los adipocitos expuestos al tratamiento HGHI, se evaluó la acumulación de triglicéridos intracelulares, mediante la captación celular de la tinción de Oil Red-O, que actúa debido a su capacidad lipofílica tiñendo de color rojo los lípidos presentes en el adipocito, específicamente a las gotas lipídicas. Como se muestra en la (Figura 1c y 2a) la lipogénesis aumentó 82,18 % en los adipocitos tratados con HGHI respecto al control y alcanzó un porcentaje del contenido de triglicéridos de 182,18%.
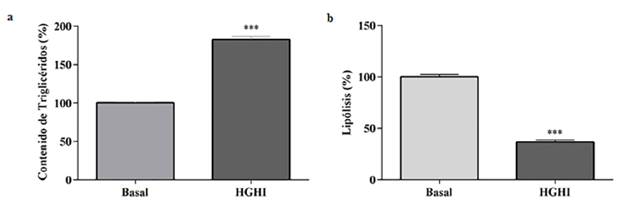
Figura 2 Evaluación de la movilización de lípidos del modelo HGHI in vitro. (a) lipogénesis, y (b) lipólisis. Los valores evaluados representan la media ± SEM. Las diferencias entre grupos se analizaron mediante ANOVA de una vía seguido de la prueba de Dunnett: *P<0,05; **P<0,01; y ***P<0,001 respecto al control.
Adicionalmente, para seguir evaluando la movilización de lípidos, se realizó la cuantificación del glicerol libre presente en el medio donde se cultivaron las células tanto tratadas con HGHI como las no tratadas, para lo cual el kit implementado mide el glicerol endógeno liberado mediante la reacción de enzimas acopladas. Específicamente, como se puede observar en la (Figura 2b), en las células tratadas con HGHI se produjo una disminución significativa en el porcentaje de lipolisis, llegando a una reducción de 36,50 % respecto al control.
Resistencia a la insulina
Con el fin de demostrar la inducción de resistencia a la insulina en los adipocitos tratados con HGHI, se determinó la relación de pAKT/AK de los adipocitos tratados y no tratados con HGHI y se encontró una disminución significativa del 54 % en el ratio de pAKT/AK de los adipocitos tratados con HGHI en relación con las células control como se observa en la Figura 3a. Además, se observó una disminución en la intensidad relativa de la proteína proinflamatoria Jun N-terminal (JNK) en comparación con el control. La intensidad relativa llegó a 0,5 en comparación con el control con un valor de 0,62; en contraste con la vía de señalización de la insulina en las células tratadas con HGHI, donde se evidenció un aumento de la intensidad relativa de 0,4; 0,8; 0,95 y 1 en la proteínas de shock térmico de 70 KDa (HSP70-marcador de estrés celular), NAD(P)H Quinone Dehydrogenase 1 (NQ01), con función de protección contra el estrés oxidativo, tirosina IRS-1 (IRS) y una diferencia estadísticamente significativa (<0.05) en la tirosina fosforilación IRS-1 (pIRS), que funcionan como sustrato para el receptor de insulina y otras tirosinas quinasas, en comparación a las células control respectivamente. Por el contrario, la proteína fosforilada proinflamatoria Jun N-terminal (pJNK-marcador de inflamación) no mostró diferencias significativas tras tratamiento con HGHI.
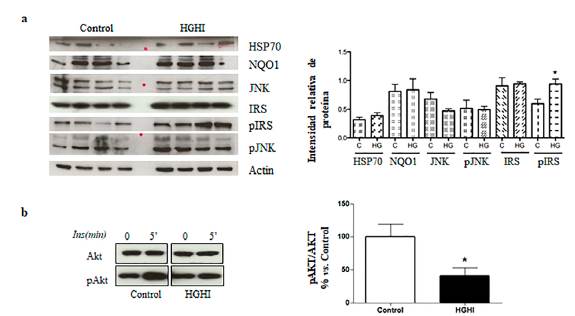
Figura 3 Señalización de insulina en modelo de HGHI (a) Western blot de HSP70, NQO1, JNK, IRS, pIRS, pJNK (imagen izquierda) e intensidad relativa de las proteínas (tabla derecha) en ausencia (control) o presencia del tratamiento HGHI (b) efectos del modelo HGHI en la vía de señalización de insulina en los adipocitos (ratios pAkt/Akt) con y sin tratamiento de HGHI. Los valores evaluados representan la media ± SEM. Las diferencias entre grupos se analizaron mediante ANOVA de una vía seguido de la prueba de Dunnett: *P<0,05; **P<0,01; y ***P<0,001 respecto al control.
Discusión
Los preadipocitos de la línea celular 3T3-L1 son ampliamente empleados en el estudio de la obesidad debido principalmente a la facilidad en el mantenimiento de cultivos y su estabilidad, al tolerar un amplio número de subcultivos en comparación a otras formas de cultivo como lo son los cultivos primarios15. Debido a esto, los adipocitos diferenciados se han utilizado ampliamente en diferentes estudios que evalúan la adipogénesis, la movilización de lípidos, para describir las especies reactivas de oxígeno (ROS) o el papel de algunas hormonas como la testosterona e inclusive se han aplicado en técnicas de silenciamiento o transfección de algunos virus16. A pesar de que, la obesidad se caracteriza por modificar la estructura de los adipocitos, en donde ocurren procesos de hiperplasia e hipertrofia, cuando se excede el equilibrio de consumo y gasto energético17. Al investigar la obesidad y sus efectos normalmente hay diversidad de modelos disponibles10-13. Sin embargo, pocos están caracterizados y no simulan el estado de obesidad patológica in vitro. Por lo anterior es necesario un modelo in vitro de hipertrofia e insulino resistencia, se propone la caracterización y uso de un modelo HGHI.
Inicialmente, sobre los resultados de viabilidad celular de los adipocitos tratados con HGHI en comparación con el modelo control de células 3T3-L1 (células no tratadas) este ensayo permitió concluir que el tratamiento HGHI no generó citotoxicidad en los adipocitos ya que los adipocitos tratados presentan una alta viabilidad, lo que demuestra una tolerancia óptima al tratamiento con una supervivencia de 238,96 % al ser expuestos al tratamiento para inducir el modelo de obesidad y resistencia a la insulina12,13. Otros estudios han realizado ensayos similares de viabilidad empleando solamente insulina a concentraciones de 106 pmol/L y han demostrado que no hay toxicidad inducida por ese tratamiento13,14 Además, teniendo en cuenta que en estados de obesidad fisiológicos se aumenta la capacidad lipogénica en los adipocitos, tal como ocurrió en los resultados obtenidos, debido a que la lipogénesis es un proceso que estimula la acumulación de triglicéridos15. Estos resultados sugieren que el modelo in vitro propuesto HGHI imita la condición fisiológica de desequilibrio energético de un adipocito hipertrófico en dónde se incrementa la acumulación de lípidos12.
Por otra parte, la adipogénesis in vitro está regulada por las concentraciones elevadas de glucosa en el medio de cultivo. Los cambios en el metabolismo lipídico durante esta etapa se acompañan de cambios morfológicos y funcionales en los adipocitos, que sufren un proceso de hipertrofia. (Figura 1d) Conjuntamente ya que en estados de hipertrofia e hiperplasia del adipocito alteran la regulación de la homeostasis energética, la cual está relacionada con la degradación o lipolisis de los triglicéridos acumulados durante la lipogénesis, evaluamos el comportamiento de la lipolisis en los adipocitos tratados con el modelo HGHI en comparación al control, encontrándose una disminución en la lipolisis del modelo patológico, lo cual era de esperarse ya que en el estado de obesidad se altera el proceso de lipolisis (Figura 2b)17. Estos resultados son similares a los obtenidos en estudios previos donde han demostrado que la disminución de la lipolisis se encuentra inducida por la activación de AMPK, debido a mecanismos dados por la hormona sensible a lipasas (HSL) o por la lipasa de triglicérido adiposo (ATGL)18.
Por otra parte, se encontró una disminución significativa de la ratio de pAKT/AK de los adipocitos tratados con HGHI en relación con las células controles como se observa en la (Figura 3a), demostrando la inducción de resistencia a la insulina en los adipocitos tratados con el tratamiento HGHI. AKT es una proteína quinasa en sí misma, y su actividad puede estimarse a partir de la fosforilación de sus sustratos. A la fecha se conoce que AKT desempeña un papel importante en las acciones metabólicas de la insulina, incluyendo la incorporación de glucosa en el músculo y el tejido adiposo a través de la translocación de GLUT-4 de compartimentos intracelulares a la membrana celular, para aumentar la captación de glucosa en la célula19. En realidad, una disminución en la relación pAkt/Akt significa que la proteína está inactiva y que probablemente disminuye la activación de la vía PI3K/Akt, debido a que la fosforilación de AKT es necesaria, pero no suficiente para su activación, ya que pAkt es la forma activa de Akt Se observó que AKT está reducida lo que desencadena en una célula insulinoresistente20,21. Existen estudios previos que evalúan varias quinasas inflamatorias en adipocitos maduros con altas concentraciones de glucosa21. Sin embargo, no se ha caracterizado un modelo en conjunto con altas concentraciones de glucosa e insulina en un solo tratamiento.
La actividad de la quinasa inflamatoria c-jun N-terminal quinasa (JNK) es elevada durante la obesidad y la diabetes y puede inducir resistencia a la insulina y disfunción de las células B, En este sentido, la expansión de los adipocitos que se produce durante la obesidad lleva asociado un proceso de inflamación crónica de baja intensidad en el que la actividad de JNK se encuentra anormalmente elevada, ya que responde a diversas señales de estrés celular activadas por citocinas, ácidos grasos libres e hiperglucemia, mediando la transición entre la obesidad y la DMT222. De manera interesante observamos una tendencia significativa a la reducción en la proteína proinflamatoria Jun N-terminal (JNK) en comparación al control, por lo que es previsible que moléculas capaces de interferir con la ruta de la JNK disminuyan la actividad de JNK. En consecuencia, la inhibición de la ruta de JNK está asociada a una mejora de la respuesta a la insulina como un mecanismo compensatorio. Esta hipótesis está apoyada por el hecho de que moléculas que normalmente están implicadas en el mantenimiento de la homeostasis celular actúan a través de sus receptores intracelulares y son capaces de inhibir la activación de la ruta de la JNK23.
Por el contrario, la autofosforilación de la proteína proinflamatoria Jun N-terminal (pJNK) no se ve afectada por la presencia del tratamiento HGHI ya que no se observan diferencias significativas en comparación a las células no tratadas con HGHI24. En contraste con la vía de señalización de la insulina en las células tratadas con HGHI, se evidenció un aumento de las proteínas de shock térmico de 70 KDa (HSP70), NAD(P)H Quinone Dehydrogenase 1 (NQ01), tirosina IRS-1 (IRS) y tirosina fosforilación IRS-1 (pIRS), respecto a las células control. La tendencia en el aumento de la HSP70, NQ01, IRS, pIRS permite percibir que el tratamiento HGHI desencadena resistencia a la insulina como mecanismo compensatorio a procesos de inflamación, mayor estrés oxidativo y estrés del retículo endoplásmico en presencia de glucosa alta25,26. Además, en investigaciones previas se ha demostrado que el estímulo con insulina genera un aumento en la fosforilación de IRS-1 (pIRS), similar a los resultados obtenidos en este estudio donde se evidencio un aumento estadísticamente significativo de pIRS (Figura 3a) lo que comprueba que este participa como mecanismo clave en la promoción de la resistencia a la insulina27,28. Lo anterior constituye un mecanismo de defensa que permite a los adipocitos adaptarse a condiciones de estrés oxidativo, inflamación y disfunción mitocondrial para aumentar su capacidad de protección y supervivencia celular frente al desarrollo de ciertas patologías, incluidas la obesidad y la resistencia a la insulina, mecanismo que también ocurre en sujetos obesos in vivo29 , 30.
Conclusiones
Se requiere un sistema de modelo de obesidad in vitro para dilucidar los mecanismos moleculares relacionados con la transducción de señales y los segundos mensajeros que participan en el proceso de señalización de insulina, por lo que, en este estudio se investigó el efecto de altas concentraciones de insulina y glucosa en células 3T3L1. Por lo anterior, los resultados demuestran la validez del modelo in vitro de obesidad al emplear un tratamiento con alta glucosa, alta insulina como una herramienta para estudiar los mecanismos de susceptibilidad a obesidad y resistencia a la insulina inducida. Este trabajo establece y caracteriza el modelo de obesidad in vitro en células 3T3L1 al usar concentraciones altas de glucosa e insulina para la simulación in vitro de la obesidad y la resistencia a la insulina de manera sencilla, económica y rápida. En estudios posteriores se podrían abordar marcadores relacionados a determinar la vía en la que se produce el aumento de la lipogénesis y la disminución de la lipolisis como AMPK o la implicación en los genes que participan en la diferenciación de adipocitos, además estudios de proteómica con el fin de determinar posibles alteraciones en la señalización de la insulina con el fin de ampliar la información de los datos obtenidos en el presente estudio.

