











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkI. INTRODUCTION
Zeolites represent a very important family of crystalline microporous aluminosilicates that allow the adsorption of water and other cations. They are characterized by a three- dimensional (3D) structure, with [SiO4][4] and [AKX][5] tetrahedra sharing corners that form cavities occupied by large ions and water molecules with great freedom of movement, which promote ion exchange and reversible dehydration. Zeotypes can be distinguished by their Si/Al ratio and the 3D cages types. Several low-silica zeotypes with Si/Al ratio of 1:1, such as zeolite Linde Type A (LTA), sodalite (SOD) and cancrinite (CAN), can be obtained from kaolin without any additional SiO2 source [15]. Selim and Maksod [6] reported the formation of zeolite X (FAU) and zeolite P (GIS) from kaolin using an additional SiO2 source. Moreover, higher Si/Al ratio zeolites as mordenite, ZSM-5 and NaY can be synthesized from kaolin and additional SiO2 source as raw materials [5,10]. Healey et al. [7] synthetized a pure JBW framework under hydrothermal conditions. The JBW structure, which is one of the least investigated zeolites, has the general formula (Na3H2O) (Si3Al3O12) [11]. The Structure Commission of the International Zeolite Association assigned the framework-type code (FTC) to the JBW structure, which is derived from the name of the type material Na-J and the initials of the author's names, Barrer and White [12], who were the pioneers in preparing this zeotype. Subsequent work referred it inappropriately as "nepheline hydrate I", due to its similarity in chemical composition with an anhydrous nepheline, although its crystalline structure is unrelated to it. Fig. 1 shows the JBW structure according to Baerlocher and McCusker [11]; the structure of the JBW framework is shown in the upper part. According to Heller-Kallai and Lapides [2], the JBW framework can be described by a zigzag chain -bold in the left middle part- running parallel to a -or using the saw chain running parallel to b (lower part)-. The repeated distance along the zigzag chain is about 5.2 Å. The two-dimensional (2D) Periodic Building Unit (PerBU) is obtained when the zigzag chains are connected along c into a layer of 6-ring (fused) chairs capped with additional zigzag chains, as shown in the left middle part. An alternative (one-dimensional) PerBU is obtained when 6-ring chairs are 2-fold connected along, forming a cylinder, as depicted in the right middle part. The cylinder wall consists of 8-rings (fused). Hansen and Fälth [13] solved the crystalline structure of the JBW structure (with the exception of a full description of the water molecules), which can be described as orthorhombic, crystallizing in the Pna21 space group, and with unit cell parameters a=14.426[1] Å, b=15.014(5) Å, c=5.224(5) Å. They synthetized the JBW structure under hydrothermal conditions at 200 °C from a sodium aluminosilicate glass in sodium hydroxide solution, although reaction products such as "nepheline hydrate", analcime and cancrinite were formed [13]. Moreover, in the past several years, numerous studies have assessed the JBW structure [12,16]. Healey and co-workers [7,8] prepared pure aluminosilicate JBW by hydrothermal reaction of kaolin in alkaline solutions, using NaOH and KOH as activating agents at 225 °C, and the Na/Rb-AlGe-JBW structure from a Na and Rb-containing reagent at 225 °C. Shimizu and Hamada [17] obtained large crystals of the JBW framework and other zeotypes using a ceramic tube as aluminosilicate source through a bulk material dissolution technique. Tripathi and Parise [18] synthesized aluminogermanate analogues of the JBW structure (Na-AlGe-JBW structure) at 140 °C from a reactant mixture containing tetramethyl ammonium hydroxide (TMAOH). Lin et al. [14] studied the synthesis of the SOD, CAN and JBW structures under hydrothermal conditions at 200 °C, using metakaolinite as starting material. Ríos' [15] experiments focused on the synthesis of a JBW type structure using metakaolinite and precipitated SiO2 as starting materials. In addition, Ríos et al. [19] synthesized several kaolinite-based zeotypes, including a nearly pure JBW-type structure obtained under hydrothermal conditions at 200 °C, using NaOH as activating agent and triethylamine (TEA) as a structure-directing agent (SDA). The synthesis of JBW-type zeolites under alkaline solutions with aerosil and aluminium triisopropoxide as starting materials, NaOH as activating agent, and phenol as an organic solvent was reported for the first time by Wei et al. [20]. The JBW-type structure is unusual because its near parts are separated by what is effectively a layer of the cristabolite structure containing non-hydrated cations [13]. According to Weller [21], the existence of this semi-condensed block helps stabilizing higher Al contents, and the JBW framework is invariably found for a Si/Al ratio of 1:1. The JBW-type structure represents a true hybrid-layered oxide/zeolite structure with alternating channels and condensed non-hydrated oxide blocks [21]. This material, obviously, has structural relationships with pillared clays [22] and the layered complex metal oxides of the CsNbTi2O7 type [23]. The JBW framework has the advantage of asymmetric pore with dimension 4.8x3.8 Å compared with 4.1x4.1 Å for LTA and 7.4x7.4 Å for FAU zeolites. This asymmetric pore may play an important role as a selective molecular sieve and also in the adsorption behavior [6]. In this work, we characterized the kaolin-based JBW framework obtained from hydrogels under hydrothermal conditions by alkaline reaction, using NaOH as activating agent.
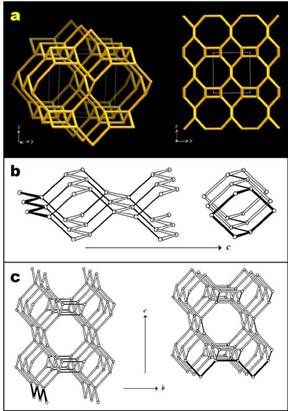
FIG. 1 (a) Structure of the JBW framework viewed along [100] (left) and projected along [100] (right).
(b) PerBU of the JBW structure viewed along a, constructed using the zigzag chain. (c) Connection mode of the JBW structure viewed along a. Adapted and modified after Baerlocher and McCusker [11].
II. EXPERIMENTAL
A. Synthesis of kaolin-based JBW framework and associated phases
Ríos [15] performed the synthesis of zeotypes, including the JBW framework, using the following procedure: A calculated amount of sodium hydroxide (NaOH) pellets was dissolved by stirring in distilled water in 150-250 ml beakers to prepare 1.33M and 3.99M NaOH solutions. Then, kaolin was added to the alkaline solutions under stirring conditions until it dissolved completely, to homogenize a reaction hydrogel with a specific molar composition. The influence of calculated amounts of structure-directing agent (SDA) in the reactant mixture was evaluated to obtain several zeotypes. The resulting hydrogels were then transferred to 65 ml polytetrafluoroethylene (PTFE) vessels (Cowie Technology Ltd) and 20 ml stainless steel autoclaves, which were heated in an oven under static conditions at 100 and 200 °C, respectively, at several reaction times. Subsequently, the vessels and autoclaves were removed from the oven and quenched in cold water to room temperature. The pH of hydrogels before and after hydrothermal synthesis was measured. The synthesis products were recovered by filtration, washed with water and dried at 80 °C overnight.
B. Characterization of the JBW framework and associated phases
The crystalline phases of the untreated kaolin and as-synthesized products were identified by X-ray powder diffraction (XRPD) and recorded in a Philips PW1710 powder diffractometer, using Cu-Ka radiation at 40 kV and 40 mA. The samples were scanned in the 26 interval 3-50°. Qualitative phase analysis was conducted using the Joint Committee of Powder Diffraction Standards (JCPDS) database and the Crystallographica Search-Match software. Data analysis was performed by the Rietveld method using the Jana-2006 software [24]. Scanning electron microscopy (SEM) was performed on a ZEISS EVO50, under the following analytical conditions: I probe 1 nA, EHT=20.00 kV, beam current of 100 uA, Signal A=SE1, WD=8.0 mm. Structural characterization was conducted by Fourier transform infrared (FT-IR) spectroscopy with a Mattson Genesis II FT-IR spectrometer for wave number range of4000400 cm-1. However, we discuss only the 1200-400 cm-1 region, which showed remarkable changes.
29Si and [27]Al magic angle spinning nuclear magnetic resonance (MAS-NMR) spectra were obtained on a Varian Unity INOVA spectrometer. 29Si MAS-NMR spectra were recorded at 59.6 MHz using 30 us acquisition time; 120 s repetition time; 5040 Hz spinning rate; 90.0° pulse angle (DP). 27Al MASNMR spectra were recorded at 78.1 MHz using 10 us acquisition time; 0.5 s recycle time; 14000 Hz spinning rate; 18.9° pulse angle (DP). The chemical shifts for [29]Si and [27]Al, respectively, were referenced to tetramethylsilane (TMS) and 1 M AlCk aqueous solution.
III. RESULTS AND DISCUSSION
The characteristic morphology of the JBW zeotype crystals (long prismatic lath-like with pseudohexagonal cross sections) was observed in the SEM micrographs [Fig. 2). Wei et al. [20] reported similar morphologies in the JBW-type zeolite obtained by phenol solvothermal synthesis. However, Hegazy et al. [25], using kaolin as starting material in the hydrothermal reaction, obtained JBW crystals with a cylindrical shape, developing spherical agglomerates.

FIG. 2 SEM micrographs of the JBW zeotype crystals obtained after hydrothermal reaction of kaolin in 3.99 M NaOH solutions at 200 oC after 168 h.
Fig. 3 illustrates the XRPD patterns of the raw kaolin and the reaction products obtained after hydrothermal transformation of kaolin in 3.99 M NaOH solutions at 200 oC for several reaction times. The transformation of kaolinite can be revealed by the formation of the JBW zeotype (with higher intensity peaks) with minor amounts of SOD and CAN, which is in agreement with Lin et al. [14], who concluded that the activation at higher temperature promotes the formation of the JBW zeotype. The reaction route allows concluding that the raw kaolin was quickly dissolved giving rise to the formation of several zeotypes, increasing the JBW-type zeolite crystallinity as temperature increased. However, the crystallinity of SOD and CAN showed a constant behavior.
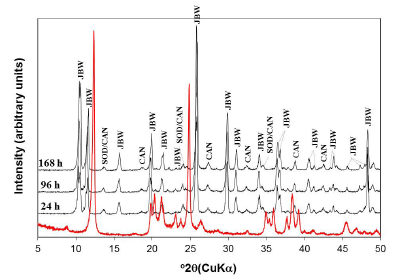
FIG. 3 XRPD patterns of the JBW-type structure obtained after treatment of kaolin (red line) in 3.99 M NaOH solutions at 200 oC for several reaction times.
To characterize the obtained JBW-type zeolite, we performed a careful analysis using the Rietveld method. As we mentioned above, the structure of this compound was originally described using an orthorhombic symmetry with cell parameters a=15.157 Å, b=8.218 Å and c=5.222 Å and space group Pmn2 1 [26]. In this sense, this model was used as a first approximation to explain the measured pattern that led to a failed result. Following the search for reported JBW-type structures, doubling the b parameter was considered according to Hansen and Fälth [13]. The next refinements considered a double cell in [10] direction. However, in this case, the refined b parameter converged in a different value, far from a simple double parameter. The results led us to consider the model proposed by Healey et al. [7], which was taken as a starting approximation to the true structure (Fig. 4). The refinements using this model allowed us to confirm that the JBW-type structure obtained by the synthesis method used in this work crystallizes in an orthorhombic system with cell parameters a=16.409(2) Å, b=14.966(2) Å and c=5.2154(5) Å and most probable space group Pna2\ t with agreement factors Rp=7.18, Rwp=10.05 and X2=1.4. These values are close to the originals reported by Healey et al. [7]: a=16.2906(2) Å, b=15.0494(2) Å and c=5.17904(6) Å. A comparison of the cell volumes, Vou=1280.8(2) and VHealey=1269.71(2), suggests an increment in the water molecules contained in the structure. This consideration allows imagining a probable Na2K[Al3Si3O12]xH2O formula for the synthesized zeolite with x~2. A comparison of the unit cell parameters calculated for several JBW zeotypes is given in Table 1, where a correlation with water content can be observed. Considering the similarities with the literature, a sodium-potassium chemical composition is not a surprise considering that the kaolinite precursor was in fact a natural mixture with other phases such illite and muscovite. However, the X-ray diffraction data did not allow us to confirm such assumption, thus, our interpretation relies on the excellent agreement with the structure calculated by Healey et al. [7] using neutron diffraction. This stoichiometry implies that two of three cation sites should be occupied by Na ions, with K ions situated on a distinct site (Fig. 4).
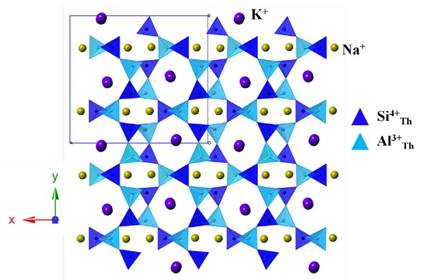
FIG. 4 Structural model used to refine the JBW-zeolite (after Healey et al. [7]). Space group Pna2 x , a = 16.409(2) Å, b = 14.966(2) Å and c = 5.2154(5) Å.
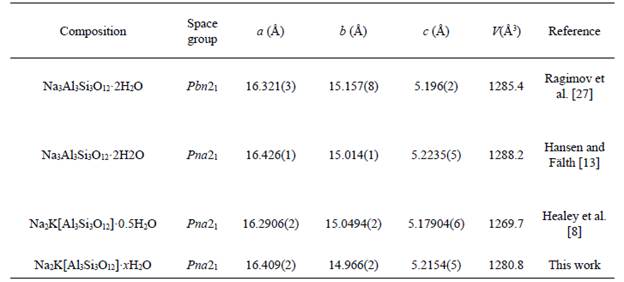
Fig. 5 shows the FT-IR spectra of the raw kaolin and the reaction products obtained after hydrothermal transformation of kaolin in 3.99 M NaOH solutions at 200 oC for several reaction times. The characteristic vibration bands (OH-stretching at 3687 and 3619 cm- 1, the surface OH bending at 1819, 1034 and 942 cm- 1, the inner OH bending at 916 cm-1, and the bands at 801, 762 and 696 cm-1) of the starting raw material (kaolin) disappeared with reaction time. In general, these bands progressively disappeared and changed in intensity and position at lower wavenumbers, which reveal that there are more Al substitution in the tetrahedral sites with NaOH acting as a structure modifier, as reported in previous studies [28,30]. The vibration bands of the synthesis products at low temperature show the asymmetric and symmetric vibration bands characteristic of a mixture of SOD and CAN in the region located between 1200 and 400 cm-1. Barnes et al. [31] summarized the reported assignments of vibration bands for SOD and CAN in this range. Simultaneously to the disappearance of the vibrations bands of the kaolin, typical zeolite bands appeared on the spectra, including the asymmetric and symmetric Al-O stretch vibrations located in the regions of 1250-950 cm-1 and 770-660 cm-1, respectively. A single vibration band at around 960-965 cm-1 can be attributed to the presence of CAN. The asymmetric stretch vibration of the Si-O-Al linkage in the SOD consists of a single band at 980 cm-1, which split into four bands at 1110, 1050, 1020, and 960 cm-1 assigned to CAN as suggested by Barnes et al. [31]. However, only the bands at 949 and 953 cm-1, assigned to SOD and CAN, respectively, are reported in this study. A weak vibration band at 754-756 cm-1 can be attributed to the presence of CAN. Several bands in the region between 760 and 650 cm-1 revealed a mixture of SOD and CAN in the reaction products. Breck [32] attributed the vibration bands in the region between 650 and 500 cm-1 to the presence of double rings (D4R and D6R) in the zeotype frameworks. In this region, the vibration bands at 619-621, 594, 554 and 507 cm-1 can be attributed to the presence of six-rings of the dehydrated region and eight-rings of the hydrated section of the JBW zeolite. The vibration bands in the region between 500 and 420 cm-1 were assigned to internal tetrahedron vibrations of Si-O and Al-O in the as-synthetized zeotypes. The range of 800-400 cm-1, which has been considered as the 'fingerprint' region for SOD and CAN in previous studies [29,31], also reveals the presence of the JBW zeolite.
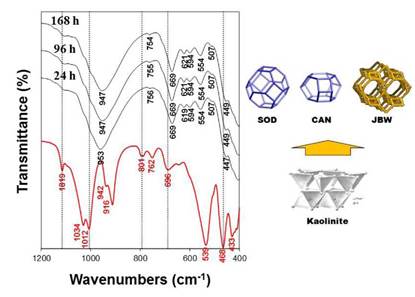
FIG. 5 FT-IR spectra of the JBW-type structure obtained after treatment of kaolin (red line) in 3.99 NaOH solutions at 200 oC during several reaction times. Kaolinite structure after Klein and Hurlbut [33]. JBW structure and sodalite (SOD) and cancrinite (CAN) cages after Baerlocher and McCusker [11].
Si-atoms in zeolite frameworks are generally coordinated to four O-atoms in Qn tetrahedrons, resulting in a five chemical environment of Si-atoms (Fig. 6a), which can be named as Si(nAl) units. The Qn notation describes the substitution pattern around a specific Si-atom, Q represents a Si atom surrounded by four O-atoms, and n, the number of Al-atoms in the second sphere of coordination or connectivity [34,35]. According to Cestari et al. [36], in these structures the Si- and Al-atoms are tetrahedrally coordinated with each other through shared oxygen atoms. 27Al is a spin 5/2 nucleus, therefore, quadrupolar, and is a high sensitivity nucleus that yields broad lines over a wide chemical shift range [34]. As a result, the signal width increases with the asymmetry of the environment, with somewhat broad lines in symmetrical environments (Fig. 6b) but very broad lines in asymmetric ones.
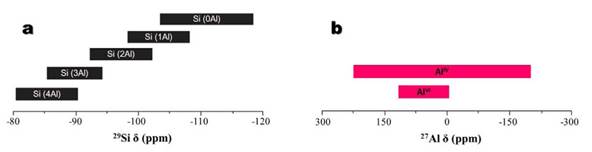
FIG. 6 Chemical shift ranges for (a) Si(nAl) chemical environment of Si-atoms by [29]Si NMR after Mafra et al. [35], and (b) [27]Al NMR after [37].
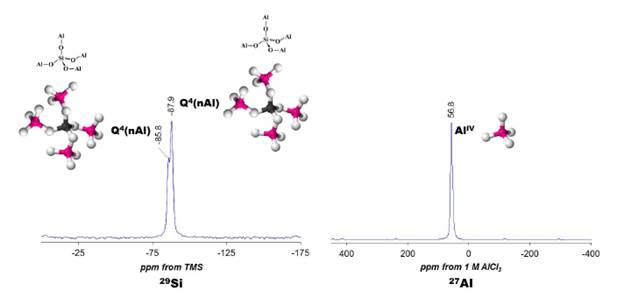
Fig. 7 illustrates the [29]Si and [27]Al NMR spectra of the JBW zeotype. The [29]Si NMR spectrum (left), which allows investigating the chemical environment of Si-atoms in the JBW structure, revealed two Si environments, with signals at -85.8 and -87.9 ppm. Similar results have been reported by other authors [7,8,14] to confirm the characteristic framework of the JBW-type structure. They attributed the smallest of the two peaks (at -85.8 ppm) to Si(1), whereas other two chemically similar Si-atoms, Si(2) and Si(3), lead to the largest of the two peaks (at -87.9 ppm). However, the weak signal at -85.8 ppm can be attributed to the contribution of Q4(4Al) sites of SOD+CAN. The [27]Al a 4-coordinated Al local environment in the JBW-type NMR spectrum (right) of the JBW framework showed structure. a single resonance at 56.8 ppm, which was assigned to a 4-coordinated Al local environment in the JBW-type structure.
IV. CONCLUSIONS
The JBW-type framework with high crystallinity was successfully synthesized by hydrothermal treatment of raw kaolin as SiO2 and Al2O3 source, in 3.99 M NaOH solutions at 200 oC, during several reaction times. The role of a SDA in the reactant mixture was evaluated to estimate how it controls the formation of several zeotypes. SOD and CAN crystallized along with the JBW zeotype. The as-synthesized zeotypes were characterized by analytical techniques such as XRPD, SEM, FT-IR and MAS-NMR. The JBW zeotype crystallized in the orthorhombic space group Pna2 1 , with unit cell parameters a=16.409(2) Å, b=14.966(2) Å and c=5.2154(5) Å. SEM analysis revealed that zeotype crystals show a long prismatic lath-like shape with pseudohexagonal cross sections. FT-IR and MAS-NMR data allowed elucidating the structural groups and the [29]Si and [27]Al environments in the as-synthesized zeolites. In general, the analytical data were consistent with experimental and theoretical reports from the literature.

