










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Facultad de Odontología Universidad de Antioquia
Print version ISSN 0121-246X
Rev Fac Odontol Univ Antioq vol.21 no.2 Medellín Jan./June 2010
REPORTE DE CASOS
Neurofibromatosis: reporte de un caso
Neurofibromatosis: a case report
Francisco Levi Duque Serna1; Omar Alberto Ramírez Usuga2
1 Odontólogo. Especialista en Estomatología. Cirugía Oral y Maxilofacial. Alta gerencia. Profesor titular Facultad de Odontología, Universidad de Antioquia. Medellín, Colombia. Dirección electrónica: flds@une.net.co
2 Odontólogo. Estudiante del posgrado de Cirugía Oral y Maxilofacial de la Facultad de Odontología, Universidad de Antioquia, Medellín, Colombia. Dirección electrónica: omarar@hotmail.com
RESUMEN
La neurofibromatosis tipo I (NF1) es una alteración autosómica dominante que se manifiesta con múltiples lesiones cutáneas, llamadas neurofibromas. Las manifestaciones orales han sido reportadas en porcentajes bajos, siendo la lengua el lugar afectado con mayor frecuencia. Presentamos el caso de una paciente afectada por neurofibromas que invaden cavidad oral y lengua que fue tratada en el servicio de Cirugía Oral y Maxilofacial del Hospital Universitario San Vicente de Paúl.
Palabras clave: neurofibroma, neurofibromatosis, enfermedad de von Recklinghausen.
ABSTRACT
Type I Neurofibromatosis (NF1) is an autosomal dominant disease that presents with multiple cutaneous lesions, named neurofibromas. Oral manifestations have been reported in low percentages, but the tongue has been the most common site. This is a report of a case of a young girl with neurofibroma which invaded the oral cavity and tongue and was treated at the Oral and Maxillofacial Surgery Service of Hospital Universitario San Vicente de Paúl.
Key words: neurofibroma, neurofibromatosis, von Recklinghausen disease.
NEUROFIBROMATOSIS
La neurofibromatosis tipo 1(NF1) descrita por primera vez por von Recklinghausen en 1884 es una enfermedad de transmisión genética autosómica dominante causada por un espectro de mutaciones al gen NF-1. Su manifestación clínica más importante es la aparición de lesiones cutáneas que pueden estar en cantidades de 500 a 1.000, principalmente en torax.1
Estas lesiones están formadas por la combinación de células de Schwann, fibroblastos, mastocitos y células perineurales. Debido a su crecimiento constante pueden comprometer estructuras óseas, vísceras, arterias y venas.2 Existe una variante clasificada como neurofibromatosis tipo 2(NF2). La NF1 se presenta aproximadamente en 1 de cada 3.000 nacimientos,3 mientras que NF2 solo se ha reportado en cerca de 1 en cada 50.000 nacimientos.4 La NF1 se caracteriza por la presencia de los siguientes signos: seis o más máculas de color “café en leche” de 0,5 cm de diámetro, múltiples neurofibromas, máculas café en leche bajo las axilas y zonas de pliegues de la piel tales como la ingle y las axilas, presencia de nódulos de Lisch (hamartomas en el iris ocular).5-8 La NF2 se conoce como neurofibromatosis acústica bilateral, se caracteriza por la presencia de múltiples tumores y lesiones en cerebro y la médula espinal.4 El primer síntoma de la enfermedad suele ser el crecimiento de tumores en los nervios auditivos que tienen como consecuencia la pérdida de audición,9 con frecuencia, esto no es evidente hasta finales de la adolescencia o principios de la adultez.1
Las manifestaciones orales de la NF1 se presentan entre el 5 y 7%. Se han reportado tumores que comprometen todos los tejidos, tanto duros como blandos, pero la lengua es el lugar más común de aparición de los neurofi bromas.10
Reportamos el caso de una paciente con diagnóstico confirmado de NF1 que presentó compromiso funcional y estético a partir de neurofi bromas que iniciaron en la región cervical izquierda y que infi ltraron los tejidos orales, principalmente la lengua.
CASO CLÍNICO
Paciente de sexo femenino de doce años, procedente de Urrao (Antioquia). El motivo de consulta principal era cefalea constante, acompañada de disfagia, mareos y con dificultad respiratoria ocasional. La paciente presentaba como antecedente quirúrgico la resección parcial de una masa en la región cérvico-occipital derecha a los tres años de edad, con diagnóstico de neurofibromatosis. No asistió a los controles programados en los últimos ocho años. Entre los antecedentes familiares de la paciente se encuentran: abuela materna con tumor en sistema nervioso central (SNC) no especificado, madre con múltiples neurofibromas en piel y un tumor en tejidos blandos de región sacra no especificado.
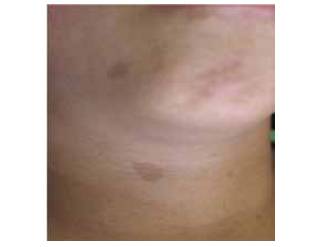
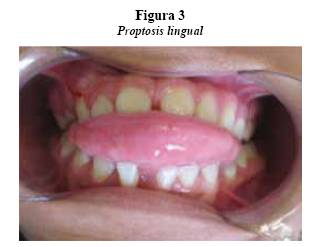
A la inspección se encontró una paciente consciente, orientada con asimetría marcada caracterizada por aumento de tamaño en región cérvico-occipital derecha, donde se observaron dos cicatrices de 85 y 25 mm, asociadas a la primera resección, a los tres años de edad. Múltiples manchas color “café en leche” en la región geniana derecha, cuello, espalda, pies y axila derecha. Presenta labios incompetentes, con lengua protruida en reposo, con amplitud de 54 mm, y que se palpa indurada. Asimétrica en toda su extensión, con mayor volumen, desde la raíz, en el lado derecho. Presenta mordida abierta dental de 9 mm y múltiples diastemas en ambos maxilares. Relación dental de clase III esquelética y patrón de prognatismo mandibular marcado (figuras 1, 2 y 3).
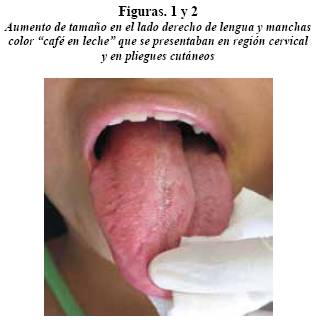
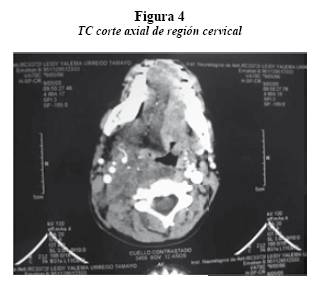
En la tomografía computarizada (TC) contrastada de cabeza y cuello se observó asimetría de los tejidos blandos del cuello del lado derecho y un franco engrosamiento de los tejidos prevertebrales desde la región de nasofaringe, de aspecto hipodenso que empuja la mucosa en sentido anterior. Además, se observó desplazamiento completo de la vía aérea hacia el lado izquierdo y del espacio vascular hacia el aspecto lateral, disminuyendo el tamaño de la vena yugular, hasta nivel de C6 aproximadamente. En el piso de boca y lengua se identificó una zona heterogénea de bajo coeficiente de atenuación en la región de la hipofaringe y a la altura del hioides. La lesión tiene densidad negativa sugestiva de componente graso y también genera desplazamiento de las estructuras vasculares. Adicionalmente, se detectaron adenopatías en las cadenas cervicales en forma bilateral, con nódulos que superan los 10 mm. Finalmente, asimetría en la posición de las glándulas salivales submandibulares, presentando la del lado derecho desplazamiento caudolateral asociado a la lesión hipodensa del piso de la lengua (figura 4).
En biopsia de la lesión, el estudio histopatológico reporta: “proliferación de forma desorganizada e irregular de unos haces de aspecto neuronal, células ahusadas, uniformes, sin atipias, separadas del tejido conectivo adyacente por bandas de tejido fibrocolágeno”, con diagnóstico de neurofi broma.
El servicio de oftalmología la evaluó, confi rmó la presencia de nódulos de Lish en ambos ojos, sin deterioro de la agudeza visual.
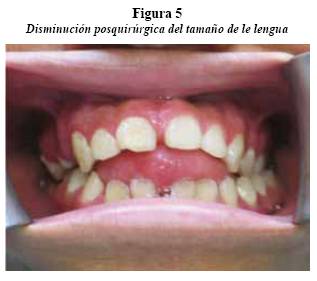
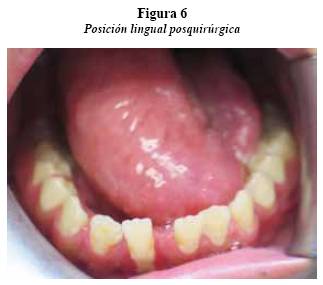
De acuerdo con estos hallazgos y con la suma de las características clínicas se llega a un diagnóstico de neurofi bromatosis tipo I. La paciente fue hospitalizada para controlar estado neurológico y evaluar compromiso de la vía aérea, fue tratada en conjunto por el Servicio de Cirugía Maxilofacial y Otorrinolaringología. Posteriormente se le realizó cirugía para la resección parcial de la masa en región cervical y glosectomía parcial. La paciente ha asistido a los controles cuatro meses después y se encuentra estable, con mejoría de los síntomas generales y muestra cicatrización adecuada en la región quirúrgica de la lengua (figuras 5 y 6).
DISCUSIÓN
La neurofibromatosis tipo I o neurofi bromatosis de von Recklinghausen, llamada de esta manera por el patólogo alemán que la describió en 1884, es un desorden genético con características autosómicas dominantes. Tiene una incidencia de 1 de cada 3.000 nacimientos alrededor del mundo,2, 3, 11, 12 lo cual la hace una de las anomalías congénitas más comunes. Este desorden tiene una penetrancia de casi el 100% pero tiene una expresión variable,13 por lo que se pueden presentar grandes variaciones en la forma de presentación incluso en una misma familia.14, 15 50% de los casos se dan de manera espontanea, es decir, a partir de mutaciones directas de células germinales.10
El diagnóstico de NF1 se basa en criterios clínicos establecidos por el Instituto de Salud Nacional de Estados Unidos en 1988.7, 8, 11 Típicamente los individuos con NF1 presentan anomalías en la pigmentación de la piel desde etapas tempranas de la vida, entre estas están las manchas “café en leche”, máculas en pliegues cutáneos y hamartomas del iris (también llamados nódulos de Lisch).5, 6 Las manchas “café en leche” son el primer signo de NF1, ocurren en 99% de los pacientes menores de un año.8 Las máculas en los pliegues cutáneos son el signos más específico de NF1, son consideradas casi patognomónicas, y se dan entre los tres y los cinco años de edad. Su causa es desconocida.7, 16
Los niños con NF1 pueden presentar, antes de los seis años, tumores gliales de bajo grado que comprometen la vía óptica, pudiendo ser gliomas o astrocitomas.13 Aunque estos normalmente no progresan, aquellos que lo hacen pueden resultar en pérdida de la visión, disfunción hipotalámica y otros síntomas de disfunción neurológica.17
Las lesiones por NF1 son generalmente tumores que pueden encontrarse en cuatro formas diferentes: cutáneas, subcutáneas, plexiformes difusa y plexiforme nodular.18, 19
Las lesiones cutáneas por NF1 aparecen comúnmente antes o durante la adolescencia y aumentan en cantidad y tamaño con la edad. Pueden variar en cantidad desde unas pocas hasta miles, con la mayor densidad de estas concentrada en el tronco. El prurito local puede estar asociado con crecimiento acelerado de los tumores y con estrés emocional, lo cual puede ser el motivo inicial de consulta de los pacientes.1, 19
Los neurofi bromas subcutáneos son similares a las presentaciones cutáneas y usualmente aparecen al inicio de la adolescencia o en la adultez temprana.19, 20 Se presentan como nódulos firmes, deprimibles, a lo largo del trayecto de nervios periféricos. Tanto las formas cutáneas como subcutáneas son benignas y no tienen riesgo de transformación maligna.1, 21
Los neurofibromas plexiformes nodulares o difusos se vuelven sintomáticos por su gran tamaño potencial y la posibilidad de estimular estructuras óseas profundas o de comprimir tejidos vecinos. A diferencia de las formas cutáneas y subcutáneas, los neurofi bromas plexiformes tienen riesgo promedio de 5% (2 a 16%13) a lo largo de la vida de transformación en tumor maligno periférico de la vaina nerviosa.1, 22, 23
Los neurofibromas plexiformes son congénitos en su mayoría y se presentan en el 30% de los pacientes con NF1. Estos tumores representan la principal causa de morbilidad y mortalidad, afectan las porciones largas de nervios periféricos y el tejido adyacente, causando malformaciones muy extendidas y complicaciones mecánicas que pueden afectar las estructuras vasculares profundas.13 De la misma manera, pacientes con NF1 presentan con más frecuencias otros tumores como los feocromocitomas y leucemias.17
El gen afectado en la NF1 se presenta en 17q.11.2,24, 25 un gen de gran tamaño que puede medir hasta 350 kb. El producto de este gen, la neurofibromina, es una proteína citoplasmática de gran tamaño con una región central que muestra una secuencia similar a la familia de las proteínas activadores de las GTPasas (GAP), las cuales están involucradas en la regulación negativa de ras.26,27 El aumento en la actividad de ras, asociado con la ausencia de neurofibromina, ha sido relacionado con la aparición de muchos de los tumores que se presentan en los casos de NF1: neurofi bromas,28 astrocitomas29 y tumores malignos periféricos de la vaina nerviosa.30
La anomalía en el gen de la neurofibromina, en combinación con el funcionamiento de otros “genes modificadores” como el p53, es una de las posibles razones en la expresión variable de esta patología aun en individuos de una misma familia.13 Recientemente se han hecho disponibles pruebas a nivel molecular para NF1. Debido al gran tamaño del gen NF1 y la falta de mutaciones específicas, se ha implementado un protocolo escalonado. Algunos expertos opinan que los criterios de diagnósticos deberían ser modificados para incluir un test molecular positivo como un criterio diagnóstico definitivo.19, 31-34
En pacientes con NF1 se deben tener en cuenta otras anomalías que pueden estar presentes. Entre las cuales están alteraciones óseas como escoliosis, displasia de huesos largos, corta estatura, osteopenia y osteoporosis.8, 35-37 También pueden presentar dificultad de aprendizaje, déficit de atención o desórdenes de hiperactividad.38-41 Los gliomas ópticos y los nódulos de Lish son patologías oftalmológicas que también deben descartarse.42-48
El diagnóstico se basa en características clínicas, para que sea confirmatorio se deben presentar dos o más de las siguientes: 1) seis o más manchas color “café en leche” que deben ser mayores de 5 mm en pacientes prepuberales, o mayores de 15 mm en pacientes postpuberales, 2) máculas en la región axilar o inguinal, 3) dos o más neurofibromas de cualquier tipo o un neurofibroma plexiforme, 4) lesiones óseas, 5) un pariente en primer grado de consanguinidad con NF1, 6) glioma óptico y 7) dos o más nódulos de Lisch.6, 7 El 97% de los pacientes con NF1 cumplirá estos criterios después de los ocho años.8 A nuestra paciente se le confirmaron seis de estos hallazgos clínicos, exceptuando glioma óptico. Los nódulos de Lisch fueron confirmados por el servicio de oftalmología, sin encontrarse cambios en la agudeza visual.
En la región oral y maxilofacial, la NF1 de tipo plexiforme tiene particular importancia. Estos tumores se originan con frecuencia de ramas del nervio trigémino e invaden tanto la cavidad oral como sus paredes laterales.49 Los cirujanos orales y maxilofaciales han reportado con frecuencia las alteraciones estéticas severas asociadas a NF1 y la dificultad para su tratamiento.10, 49-55
Friedrich49 enumeró las alteraciones en la región maxilofacial asociadas a NF1, entre estas están la elongación y adelgazamiento de los procesos articular y coronoides, acortamiento de la rama, dientes retenidos e irregularidades del reborde marginal mandibular. Shapiro10 en un estudio tiene entre sus hallazgos neurofibromas orales, papilas fungiformes aumentadas en volumen, lesiones intraóseas que afectaban principalmente el hueso mandibular, canal alveolar inferior amplio, foramen mandibular ampliado. Otros autores han asociado la NF1 con aplasia o agenesia de segundos molares o con otras alteraciones en la cantidad de dientes presentes.49
El tratamiento de esta anomalía consiste antes que todo en la concientización del paciente y su familia de las características de la enfermedad, de su transmisión genética y de las limitaciones de su tratamiento.11 Los neurofibromas, en cualquiera de sus tipos, solo pueden ser tratados de manera quirúrgica, buscando mejoría en el aspecto cosmético y para evitar un déficit funcional.1, 8, 19 Actualmente no existe manejo farmacológico disponible, pero se están adelantando estudios con diferentes tipos de fármacos que tratan de controlar el crecimiento de los tumores, mejorar el aspecto de las manifestaciones cutáneas o disminuir la afección cognitiva que con frecuencia se da en compañía de esta patología.56-67
Los conocimientos genéticos de la función del gen afectado en la NF1 direccionan los estudios clínicos actuales hacia el desarrollo de inhibidores de ras que podrían disminuir la expresión fenotípica de este tipo de anomalías.13
REFERENCIAS
1. Levine SM, Levine E, Taub PJ, Winberg H. Electrosurgical excision technique for the treatment of multiple cutaneous lesions in neurofibromatosis type I. J Plast Reconstr Aesthet Surg 2008; 61: 958-962. [ Links ]
2. Viskochil D. Genetics of neurofibromatosis 1 and the NF1 gene. J Child Neurol 2002; 17: 562-570. [ Links ]
3. Riccardi M. Neurofibromatosis: past, present and future. N Engl J Med 1991; 324: 1283-1285. [ Links ]
4. Evans DGR, Huson SM, Donnai D, Neary W, Blair V, Newton V et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness and confirmation of maternal transmission effect on severity. J Med Genet 1992; 29: 841-846. [ Links ]
5. Young H, Hyman S, North K. Neurofibromatosis 1: clinical review and exceptions to the rules. J Child Neurol 2002; 17: 613-621. [ Links ]
6. DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000; 105: 608-614. [ Links ]
7. Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997; 278: 51-57. [ Links ]
8. DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000; 105: 608-614. [ Links ]
9. MacCollin M, Mautner V-F. The diagnosis and management of neurofibromatosis 2 in childhood. Semin Pediatr Neurol 1998; 5: 243-252. [ Links ]
10. Shapiro SD, Van Dis ML, Langlais RP, Young RS. Neurofibromatosis: oral and radiographic manifestations. Oral Surg 1984; 58: 493-498. [ Links ]
11. National Institutes of Health Consensus Development Conference. Neurofibromatosis. Conference statement. Arch Neurol 1988; 45: 575-578. [ Links ]
12. Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in southeast Wales. Brain 1988; 111: 1355-1381. [ Links ]
13. Suárez C, Rodrigo JP, Ferlito A, Cabanillas R, Shaha A, Rinaldo A. Tumours of familial origin in the head and neck. Oral Oncology 2006; 42: 965-978. [ Links ]
14. North K. Neurofibromatosis type 1: review of the first 200 patients in an Australian clinic. J Child Neurol 1993; 8: 395-402. [ Links ]
15. Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet 1993; 53: 305-313. [ Links ]
16. De Schepper S, Boucneau J, Lambert J, Messiaen L, Naeyaert JM. Pigment cell-related manifestations in neurofibromatosis type 1: an overview. Pigment Cell Res 2005; 18: 13-24. [ Links ]
17. Reynolds RM, Browning GGP, Nawroz I, Campbell IW. Von Recklinghausen's neurofibromatosis: neurofibromatosis type 1. Lancet 2003; 361: 1552-1554. [ Links ]
18. Lott IT, Richardson EP Jr. Neuropathological findings and the biology of neurofibromatosis. Adv Neurol 1981; 29: 23-32. [ Links ]
19. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol 2009; 61: 1-14. [ Links ]
20. Page PZ, Page GP, Ecosse E, Korf Br, Leplege A, Wolkenstein P. Impact of neurofibromatosis 1 on quality of life: a crosssectional study of 176 American cases. Am J Med Genet A 2006; 140: 1893-1898. [ Links ]
21. Reed N, Gutmann DH. Tumorigenesis in neurofibromatosis: new insights and potential therapies. Trends Mol Med 2001; 7: 157-62. [ Links ]
22. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. J Med Genet 2002; 39: 311-314. [ Links ]
23. Friedman JM, Gutmann DH, MacCollin M, Riccardi VM, editores. Neurofibromatosis: phenotype, natural history, and pathogenesis. 3.ª ed. Baltimore (MD): Johns Hopkins University Press; 1999. [ Links ]
24. Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990; 62: 187-192. [ Links ]
25. Cawthon RM, Weiss M, Xu G, Viskochil D, Culver M, Stevens J et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure and point mutations. Cell 1990; 62: 193-201. [ Links ]
26. Ballester R, Marchuk DA, Boguski M, Saulino AM, Letcher R, Wigler M et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990; 63: 851-859. [ Links ]
27. Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990; 63: 843-849. [ Links ]
28. Sherman LS, Atit R, Rosenbaum T, Cox AD, Ratner N. Single cell Ras-GTP analysis reveals altered ras activity in a single population of neurofibroma Schwann cells but not fibroblasts. J Biol Chem 2000; 275: 30740-30745. [ Links ]
29. Lau N, Feldkamp MM, Roncari L, Loehr AH, Shannon P, Gutmann DH et al. Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J Neuropathol Exp Neurol 2000; 59: 759-767. [ Links ]
30. Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in tumour cells from type 1 neurofibromatosis patients. Nature 1992; 356: 713-715. [ Links ]
31. Kluwe L, Siebert R, Gesk S, Friedrich RE, Tinschert S, Kehrer-Sawatzki H et al. Screening 500 unselected neurofibromatosis1 patients for deletions of the NF1 gene. Hum Mutat 2004; 23: 111-116. [ Links ]
32. Leppig KA, Kaplan P, Viskochil D, Weaver M, Ortenberg J, Stephens K. Familial neurofibromatosis 1 microdeletions: cosegregation with distinct facial phenotype and early onset of cutaneous neurofibromata. Am J Med Genet 1997; 73: 197-204. [ Links ]
33. De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet 2003; 72: 1288-1292. [ Links ]
34. Upadhyaya M, Huson SM, Davies M, Thomas N, Chuzhanova N, Giovannini S et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet 2007; 80: 140-151. [ Links ]
35. Stevenson DA, Zhou H, Ashrafi S, Messiaen LM, Carey JC, D'Astous JL et al. Double inactivation of NF1 in tibial pseudoarthrosis. Am J Hum Genet 2006; 79: 143-148. [ Links ]
36. Lammert M, Kappler M, Mautner VF, Lammert K, Storkel S, Friedman JM et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int 2005; 16: 1161-1166. [ Links ]
37. Schindeler A, Little DG. Recent insights into bone development, homeostasis, and repair in type 1 neurofibromatosis (NF1). Bone 2008; 42: 616-622. [ Links ]
38. Levine TM, Materek A, Abel J, O'Donnell M, Cutting LE. Cognitive profile of neurofibromatosis type 1. Semin Pediatr Neurol 2006; 13: 8-20. [ Links ]
39. North K. Neurofibromatosis type 1. Am J Med Genet 2000; 97: 119-127. [ Links ]
40. Goh WH, Khong PL, Leung CS, Wong VC. T2-weighted hyperintensities (unidentified bright objects) in children with neurofibromatosis 1: their impact on cognitive function. J Child Neurol 2004; 19: 853-858. [ Links ]
41. Moore BD, Slopis JM, Schomer D, Jackson EF, Levy BM. Neuropsychological significance of areas of high signal intensity on brain MRIs of children with neurofibromatosis. Neurology 1996; 46: 1660-1668. [ Links ]
42. Lewis RA, Riccardi VM. Von Recklinghausen neurofibromatosis: incidence of iris hamartomas. Ophthalmology 1981; 88: 348-354. [ Links ]
43. Huson S, Jones D, Beck L. Ophthalmic manifestations of neurofibromatosis. Br J Ophthalmol 1987; 71: 235-238. [ Links ]
44. Zehavi C, Romano A, Goodman RM. Iris (Lisch) nodules in neurofibromatosis. Clin Genet 1986; 29: 51-55. [ Links ]
45. Flueler U, Boltshauser E, Kilchhofer A. Iris hamartomata as diagnostic criterion in neurofibromatosis. Neuropediatrics 1986; 17: 183-185. [ Links ]
46. Listernick R, Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. J Pediatr 1989; 114: 788-792. [ Links ]
47. Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr 1994; 125: 63-66. [ Links ]
48. Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol 2007; 61: 189-198. [ Links ]
49. Friedrich RE, Giese M, Schmelzle R, Mautne VF, Scheuer HA. Jaw malformations plus displacement and numerical aberrations of teeth in neurofibromatosis type 1: a descriptive analysis of 48 patients based on panoramic radiographs and oral findings. J Cranio Maxillo Fac Surg 2003; 31: 1-9 [ Links ]
50. Friedman MM. Neurofibromatosis of bone. Am J Roentgenol 1944; 51: 623-630. [ Links ]
51. Rushton MA. Neurofibromatosis affecting the jaws. Am J Orthodont Oral Surg 1944; 30: 790-792. [ Links ]
52. Freeman MJ, Standish SM. Facial and oral manifestations of familial disseminated neurofibromatosis. Oral Surg 1965; 19: 52-59. [ Links ]
53. Koblin I, Reil B. Changes in the facial skeleton in cases of neurofibromatosis. J Maxillofac Surg 1975; 3: 23-27. [ Links ]
54. Gupta SK, Kaur S, Sharma OP. Craniofacial neurofibromatosis: a roentgen profile. Australasian Radiol 1984; 28: 97-105. [ Links ]
55. D'Ambrosio JA, Langlais RP, Young RS. Jaw and skull changes in neurofibromatosis. Oral Surg 1988; 66: 391-396. [ Links ]
56. Babovic-Vuksanovic D, Widemann BC, Dombi E, Gillespie A, Wolters PL, Toledo-Tamula MA et al. Phase I trial of pirfenidone in children with neurofibromatosis 1 and plexiform neurofibromas. Pediatr Neurol 2007; 36: 293-300. [ Links ]
57. Babovic-Vuksanovic D, Ballman K, Michels V, McGrann P, Lindor N, King B et al. Phase II trial of pirfenidone in adults with neurofibromatosis type 1. Neurology 2006; 67: 1860-1862. [ Links ]
58. Widemann BC, Salzer WL, Arceci RJ, Blaney SM, Fox E, End D et al. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatos type I and plexiform neurofibromas. J Clin Oncol 2006; 24: 507-516. [ Links ]
59. Franz DN, Leonard J, Tudor C, Chuck G, Care M, Seturaman G et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 2006; 59: 490-498. [ Links ]
60. Wienecke R, Fackler I, Lisenmaier U, Mayer K, Licht T, Kretzler M. Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis 2006; 48: 27-29. [ Links ]
61. Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X et al. Nf1-dependent tumors require a microenvironment containingNf1± and c-kit-dependent bone marrow. Cell 2008; 135: 437-448. [ Links ]
62. Yoshida Y, Sato N, Furumura M, Nakayama J. Treatment of pigmented lesions of neurofibromatosis 1 with intense pulsed-radio frequency in combination with topical application of vitamin D3 ointment. J Dermatol 2007; 34: 227-230. [ Links ]
63. Nakayama J, Kiryu H, Urabe K, Matsuo S, Shibata S, Koga T et al. Vitamin D3 analogues improve cafe´ au lait spots in patients with von Recklinghausen's disease: experimental and clinical studies. Eur J Dermatol 1999; 9: 202-206. [ Links ]
64. Nakayama J, Matsuo S, Rikihisa W, Hori Y. Inhibitory effect of a new vitamin D3 analogue, 22-oxacalcitriol, on the growth of neurofibroma cells xenografted into nude mouse skin in vivo. Eur J Dermatol 1997; 7: 475-479. [ Links ]
65. Lammert M, Friedman JM, Roth HJ, Friedrich RE, Kluwe L, Atkins D et al. Vitamin D deficiency associated with number of neurofibromas in neurofibromatosis 1. J Med Genet 2006; 43: 810-813. [ Links ]
66. Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW et al. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol 2005; 15: 1961-1967. [ Links ]
67. Krab LC, de Goede-Bolder A, Aarsen FK, Pluijm SM, Bouman MJ, van der Geest JN et al. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: a randomized controlled trial. JAMA 2008; 300: 287-294. [ Links ]
RECIBIDO: MARZO 31/2009 - ACEPTADO: ENERO 26/2010
CORRESPONDENCIA
Francisco Levi Duque S.
Facultad de Odontología
Universidad de Antioquia
Medellín, Colombia
Correo electrónico: flds@une.net.co
Cómo citar este artículo:
Duque FL, Ramírez OA. Neurofibromatosis: reporte de un caso. Rev Fac Odontol Univ Antioq 2010; 21(2): 218-225.
Duque FL, Ramírez OA. Neurofibromatosis: a case report. Rev Fac Odontol Univ Antioq 2010; 21(2): 218-225.

