











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Dental enamel alterations visible during the eruption of teeth are structural anomalies with a genetic or acquired origin. They develop before teeth eruption and that is why the clinical and radiographic evaluation is critical in diagnosing, characterizing, and investigating the etiology of these anomalies. When the enamel alteration affects a group of teeth and the mineralization period is identical, it is necessary to consider the presence of an environmental or systemic toxicity.1)
Amelogenesis imperfecta (AI) consists of a genetic alteration of the normal enamel structure, which is defective because of the inadequate differentiation of ameloblasts in a process consisting of several stages: pre-secretion, secretion, and maturation2 (Figure 1).
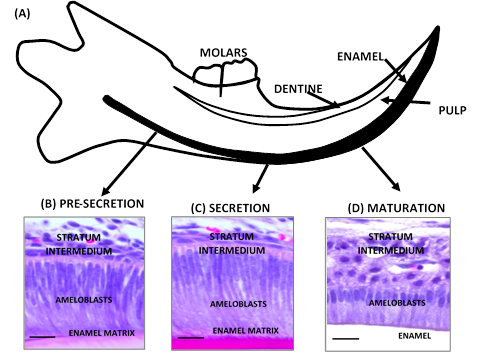
(A) Scheme of mouse incisor in continuous growth. Cross section. (B, C, D) Histology of incisor of CD1 wild mouse (3 months). (B) during the pre-secretion stage, the pre-ameloblasts lengthen and their nucleus and mitochondria migrate to the stratum intermedium. (C) in the secretion stage, the secretory ameloblasts are very elongated cells with an oval nucleus in proximal position and a well-developed area of medial and distal secretion. The Tomes’process is formed. (D) in the maturation stage, ameloblasts undergo a cyclic transformation due to the elimination of developing enamel proteins to provide mineral ions that contribute to the growth of enamel crystals. Black bar 50 µm. Source: Simancas V, Natera A, Acosta MG
Figure 1 Morphology of the process of dental enamel formation
Amelogenesis imperfecta may exist in isolation or associated to a clinical condition as part of a syndrome. Its inheritance pattern is autosomal dominant, autosomal recessive, linked to the X chromosome, or sporadic, affecting both deciduous and permanent dentition. The prevalence of AI varies from 1/700 to 1/14000 depending on the study population.3 The classification presented by Witkop,4 based on phenotype, radiographic appearance, and transmission mode is the most used currently. In fact, three large categories have been reported according to enamel defect: hypoplastic, hypocalcified and hypomaturation (Figure 2). Several forms may exist in the same patient and even in the same tooth.3
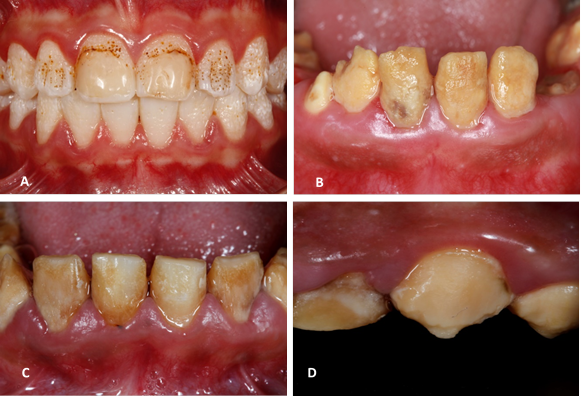
(A) Hypoplastic AI: dental enamel with thin, yellow-brown ditches or depressions (B) Hypomaturation AI: dentine is soft and rough. It shows a mottling with snowflakes appearance. (C, D) hypocalcified AI: yellow-brown soft, friable dental enamel. Note the early enamel loss. Source: Simancas V, Natera A, Acosta MG.
Figure 2 Variation of dental phenotype in amelogenesis imperfecta
Clinically, AI appears as loss of dental structure, yellow, grey or brown teeth, and dental hypersensitivity. Gingival alterations, dental eruption alterations, and taurodontism are also frequent. This condition is quite complex, and may produce social, aesthetic, and functional problems that are not easy to diagnose and treat.5 Due to the importance of AI as a clinical condition frequently found in patients of all ages, this article aims to describe the genes and mutations involved in non-syndromic AI, the proteins encoded by these genes and their functions, according to current scientific evidence.
MATERIALS AND METHODS
An electronic literature search was conducted in the Medline (PubMed), EBSCOhost and Scopus (Science Direct) databases using the following keywords: amelogenesis imperfecta, genes AND/OR amelogenesis imperfecta, and syndrome AND/OR amelogenesis imperfecta. Full articles in English, accessible in PDF and produced from the year 2000 until December 2017 were obtained as inclusion criteria. No age or sex restrictions were used. Dissertations, newspapers, conferences, news, commentaries, and editorials were excluded.
The articles used in this literature review were collected and stored using the Zotero software, which also allowed to eliminate duplicate bibliographic references. The articles were independently evaluated by two reviewers according to the above criteria. Finally, a screening was conducted applying the criteria to the articles found in the databases, and 1,573 articles were preselected; of these, 63 met the inclusion requirements and were therefore analyzed and discussed (Table 1).
Table 1 Summary of the approach used for the computerized literature search
| Keywords | Medline (PubMed) | EBSCOhost | Science Direct (Elsevier) |
|---|---|---|---|
| Amelogenesis imperfecta | 262 | 400 | 219 |
| Gene and/or Amelogenesis imperfecta | 138 | 194 | 142 |
| Syndrome and/or Amelogenesis imperfecta | 53 | 92 | 73 |
Source: Simancas V, Natera A, Acosta MG
RESULTS
Below is a description of the genes involved in non-syndromic hypoplastic, hypocalcified, and hypomaturation AI.
Genes involved in non-syndromic hypoplastic amelogenesis imperfecta
1. AMELX
The AMELX gene (OMIM 300391, amelogenin) has 7 exons and is located on the X chromosome in position Xp22.2. It encodes amelogenin, a protein rich in proline, glutamine, leucine, and histidine, and constitutes nearly 90% of the enamel organic matrix.6 Amelogenine is phosphorylated, highly hydrophobic, and relatively basic, and self-assembles in spherical aggregates known as amelogenin nanospheres. These nanospheres have an affinity for hydroxyapatite, controlling the direction of crystals in enamel and avoiding electrostatic interactions.7
The AMELX transmission mode is given by the X chromosome. Mutations in this gene cause X-linked AI (MIM # 301200).8 In females it appears as a lionization phenomenon (random inactivation of one of the two X chromosomes) responsible for an enamel with healthy and discolored stripes at the same time. In males there is a copy of AMELX as AMELY (OMIM 410000) (amelogenin) located in locus Yp11.2, responsible for encoding 10% of amelogenin.9 In the presence of AMELX gene mutation there can be a hypoplastic or hypomaturation AI in both deciduous and permanent dentition. Deletions and variants in the N-terminus cause hypomaturation AI with focal hypoplasia. On the other hand, mutations in the signal peptide and towards the C-terminus portion are responsible for a hypoplastic AI.10 Recently, Brookes et al11 pointed out that AI associated to the p.Tyr64His mutation of the AMELX gene induces stress of the endoplasmic reticulum followed by apoptosis of ameloblasts. This cellular event causes an interruption of the secretory pathway of ameloblasts that form dentin, favoring the onset of AI. However, further studies are needed to accurately elucidate the dental alterations induced by mutations of the AMELX gene.12
2. AMBN
The AMBN gene (OMIM 601259, ameloblastin) consists of 13 exons and is located on chromosome 4, position 4q13.3. It encodes for ameloblastin, the second most abundant enamel matrix protein during amelogenesis. AMBN is rich in glycine, leucine and proline, is located in the Tomes process, but has also been detected in pre-odontoblasts.13 Ameloblastin is rapidly partitioned after the secretory stage; its fragments are incorporated in the prisms and works to avoid the fusion between prisms and the interprismatic substance.
AMBN participates in the differentiation and proliferation of ameloblasts, as well as in the extracellular signaling that induces the differentiation of osteoblasts and cell adhesion.14-16 Poulter et al17 and Prasad et al1 recently reported two mutations of the AMBN gene in patients with AI. The first is a deletion in exon 6, reducing the protein amino acids from 447 to 368; clinically, an aprismatic thin enamel was found. The second mutation produced the loss of exon 7. This exon encodes a domain involved in the interaction of heparin and fibronectin, which are essential for the interaction of AMBN with epithelial cells.16-18
3. ENAM
The ENAM gene (OMIM 606585, enamelin) has 9 exons and is located on chromosome 4, position 4q13.3. It encodes for enamelin, a protein responsible for the nucleation and elongation of hydroxyapatite crystals. It expresses during the secretory stage mainly and degrades quickly from its terminal carboxyl end after its secretion by proteases, producing enamelins of a lower molecular weight that are usually found in the prisms and interprismatic substance.19,20
Wang et al21 recently reported two heterozygous mutations in the ENAM gene (c.406_407insTCAAAAAAGCCGACCACAA, p.K136Ifs*16; c. 139delA, p.M47Cfs*11). Clinically, a hypoplastic AI with horizontal veins and loss of enamel substance was observed. Saymen et al22 indicated two new heterozygous mutations of the ENAM gene: c.454G>T p.Glu152* and c.358C>T p.Gln120*. They also found that several individuals of the two analyzed families, despite having the same mutation as the affected individuals, presented incomplete penetrance.
4. LAMB3
The LAMB3 gene (OMIM 150310, laminin beta-3) is composed of 23 exons and is located on chromosome 1, position 1q32.2. This gene encodes a protein known as laminin beta-3, a subunit of laminin 5. It acts at the transmembrane level (in the basal membrane) and participates in cell growth and adhesion.21 The enamel of patients with a mutation of the LAMB3 gene shows speckles of varying extension “in a thimble from” on the surface of some or all teeth and in areas of vertical coloration.23-25
Lee et al24 identified mutations of LAMB3 in two families. In family 1, they described a mutation (c.3357 _3358insC) located 25 base pairs (bp) from the cutting and splice site of exon 22. In family 2, the mutation (c.3463_3475delGAGCAGATCCGTG), located in exon 23, produced a truncated protein of 50 amino acids. In both families there was a generalized hypoplastic AI in both deciduous and permanent dentition of the evaluated members. Poulter et al25 identified a frameshift mutation at the C-terminus portion of LAMB3. This modification was responsible for alterations in the secretory-stage amelogenesis and, consequently, an inadequate adhesion of ameloblasts to the secreted enamel matrix.
Kim et al26 reported two mutations: a deletion of 8 bp (c.3446_3453del GACTGGAG) that produced the p.Gly 1149Glufs*8 frameshift and a substitution of 2 bp (c.C3431A, p.Ser1144*) that produced the presence of a premature termination codon, shortening the protein that should have been translated. Clinically, patients show irregular hypoplastic areas and multiple cusps severely affected by AI.
5. LAMA3
The LAMA3 gene (OMIM 600805, laminin alpha-3) consists of 75 exons and is located on chromosome 18, position 18q11.2. This gene encodes for laminin alpha-3. Mutations of this gene are related to syndromic and non-syndromic hypoplastic AI. Only two mutations of the LAMA3 gene related to non-syndromic AI have been reported27 the first is located in exon 19 (c.2377C4T; p.Arg793Ter), while the second, previously described by Yuen et al,28 is located in exon 33 (c.4484C4T; p. Ala1495Val). In both affected families, there was a hypoplastic AI in the absence of clinical dermatological signs.
6. ACPT
The ACPT gene (OMIM 606362, acid phosphatase, testicular) contains 11 exons and is located on chromosome 19, position 19q13.33. It encodes an enzyme capable of hydrolyzing orthophosphoric acid esters in acidic conditions. By immunohistochemical analysis, it has been shown that ACPT is located in secretory ameloblasts, follicular cells, and osteoblasts.29 Indeed, Choi et al30 suggested that ACPT is capable of causing differentiation and mineralization of odontoblasts by supplying phosphate during dentin formation.
Seymen et al29 identified 6 families with biallelic mutations of the ACPT gene. Three homozygous mutations (c.713C>T; p.Ser238Leu, c.331C>T; p.Arg111Cys and c.226C>T; p.Arg76Cys) and two compound heterozygous mutations (c.382G> C; p.Ala128Pro and 397G> A; p.Glu133Lys) were reported. In addition, there were alterations in the sizes and lateral chains of the amino acids of the protein, which limit their accessibility to the catalytic nucleus and interfere with their homodimerization. Smith et al31 described two homozygotic mutations (c.428C>T; p. T143M and c.746C>T; p. P249L) that were responsible for generalized hypoplastic AI. Both studies reported the role that ACPT can play in the secretory stage during amelogenesis, since the analyses showed the existence of a decrease in enamel (when it was present) but at the same time showed a well mineralized enamel.
Genes involved in non-syndromic hypocalcified amelogenesis imperfecta
1. FAM83H
The FAM83H gene (OMIM 611927, family with sequence similarity 83, member H), composed of 5 exons, is located on chromosome 8, position 8q.24.3. It encodes for the intracellular FAM83H protein that participates in the differentiation of pre-ameloblasts in functional ameloblasts and in the mineralization process of enamel matrix. Its maximum expression is found in secretory ameloblasts and the minimum in the ripening stage.32,33) Its mode of transmission is autosomal dominant, and the resulting AI is hypocalcified, either localized or generalized. Clinically, deciduous and permanent teeth are affected and show mineralization defects characterized by a rough and porous dentin.34,35
Xin et al34 and Lee et al36 showed that mutations of the FAM83H gene alter the location of the protein, presenting a higher concentration within the nucleus rather than in its cytoplasmic location. Kuga et al37 showed that FAM83H regulates the organization of the cytoskeleton and maintains the formation of desmosomes. The authors suggest that, in the case of an AI resulting from an alteration of the FAM83H gene, there is a disorganization of the cytoskeletal keratin with a subsequent alteration of the desmosomes at the ameloblastic level.
2. C4ORF26
The C4ORF26 gene (OMIM 614829, chromosome 4 open reading frame 26) is composed of 2 exons and is located on chromosome 4, position 4q21.1. It encodes a protein rich in proline of the extracellular matrix containing a signal peptide with two highly conserved motifs and ten sites destined for phosphorylation. Based on the amino acids sequence, it has been estimated that C4ORF26 belongs to the family of phosphoproteins and, according to Parry et al,38 this protein promotes the crystallization of hydroxyapatite, supporting the growth of crystals after the phosphorylation of the C-terminus region.
The mode of transmission is autosomal recessive and the phenotype corresponds to a hypocalcified AI in deciduous and permanent teeth, showing a brownish yellow coloration, premature wear, and sensibility problems.2,38
3. SLC24A4
The SLC24A4 gene (OMIM 609840, solute carrier 24 A4) is composed of 17 exons and is located on chromosome 14, position 14q32.12. It encodes a protein that functions as an ion exchanger (calcium/sodium/potassium dependent ion carrier). This protein has highly conserved hydrophobic regions (alpha-1 and alpha-2) that interact in the ions bonding located at the transmembrane level.39,40 It expresses in the maturation ameloblasts and it has been suggested that it is responsible for the active transportation of Ca2+ ions from the ameloblasts to the enamel matrix during the maturation stage.41-43
Missense mutations have been described in this gene affecting the alpha regions and the cytoplasmic domain, as well as multi-exonic deletions.40,43,44 This gene expresses in a wide range of tissues, such as brain, aorta, lung, and thymus. Sulem et al45 add that it is involved in the pigmentation of eyes and hair.
4. ITGB6
The ITGB6 gene (OMIM 147558, integrin Β-6 chain) has 15 exons and is located on chromosome 2, position 2q24.2. It encodes for an integrin located in epithelial cells. This protein participates in the interactions between cells and MEC cells, facilitating the interaction with the cytoskeleton.46 Wang et al47 suggest that ITGB6 is predominantly located in the ameloblasts of the maturation stage, and would play an essential role in fibronectin receptors and in the activation of matrix metalloproteinase 20 (MMP-20) or enamelysin.
Clinically, it presents a hypoplastic AI with varying enamel compromise or a hypocalcified AI with loss of enamel substance and exogenous discolorations such as those described by Wang et al47 and Poulter et al.48 Recently, Ansar et al49 reported the missense mutation c.898G>A; p.Glu300Lys of the ITGB6 gene in a Pakistani consanguineous family, in which, in addition to a rough enamel with discolorations, intellectual disability and alopecia were reported. Its mode of transmission was autosomal recessive.
5. AMTN
The AMTN gene (OMIM 610912, amelotin) contains 9 exons and is located on chromosome 4, position 4q13.3. It encodes a protein rich in proline, leucine, threonine, and glutamine, secreted by ameloblasts in maturation stage during enamel formation. Barlette and Simmer50 estimated that AMTN forms aggregates that mediate the bonding between maturation ameloblasts and mineralized enamel. An expression of AMTN in the junctional epithelium has also been reported.51,52
Smith et al53 described the first mutation of the AMTN gene in a Costa Rican family. Mutation c.54 + 1347_330 + 98delinsCTCA consisted of a deletion of 8,678 pb covering exons 3-6 of the AMTN gene, accompanied by an insertion of 4 bp (chr4: 71,385,896-71,394,573delinsCTCT; GRCh37). The phenotypic analysis showed a hypocalcified AI with the enamel presenting a weak mineral density and an altered structure in its entire extension.
Genes involved in non-syndromic hypomaturation amelogenesis imperfecta
1. MMP20
The MMP20 gene (OMIM 604629, matrix metalloproteinase, enamelysin) has 10 exons and is located on chromosome 11, position 11q22.2. It encodes for enamelysin, a protein involved in cell motility and organic matrix degradation during the enamel maturation phase. The action of MMP20 directs the morphology of hydroxyapatite crystals and induces the increase in thickness of the hydroxyapatite crystals of dentin.54,55 Guan et al56 concluded that MMP20 can act on the extracellular domains of cadherins that allow cell-cell interactions as part of the adherent joints in the movement of ameloblastic cells. This influences amelogenesis since ameloblasts should be arranged in a synchronized manner to form a normal ameloblastic structure.
The inheritance mode is autosomal recessive and the phenotype is a pigmented hypoplastic or hypomaturation AI. Homozygotic (c.323A>G, c.954-2A>T, c.6787T>A) and compound heterozygous mutations (c.567T>C, c.910G>A, c.126+6T>G, c.954-2A>T, c.389C>T, c.954-2A>T and c.540T>A) have been reported as responsible for a porous, opaque, yellow enamel with severe wear and occasional mottling.57,58
2. KLK4
The KLK4 gene (OMIM 603767, kallikrein 4) is composed of 5 coding exons and is located on chromosome 19, position 19q13.41. It encodes a serine protease that is expressed and secreted by ameloblasts in the maturation stage during amelogenesis. This serine protease participates in the nucleation and mineralization of enamel.59 Yamakoshi et al60 showed that MMP20 can activate the newly secreted KLK4 and in turn KLK4 can inactivate the MMP20, an event that explains the change in protein activity during the maturation stage. While contributing to the elimination of enamel proteins, KLK4 allows the growth of hydroxyapatite crystals.61
Smith et al62 reported the autosomal recessive mutation c.632delT, p. L211Rfs*37 of the KLK4 gene in five families in Pakistan, which produced a premature stop codon and consequently an improper protein folding due to the alteration of three disulfide links. A low-mineralized enamel and a lesser amount of calcium and phosphorus were reported, as well as more nitrogen, compared to a control dentin.
3. WDR72
The WDR72 gene (OMIM 613214, protein 72 with repeated WD) consists of 20 exons and is located on chromosome 15, position 15q21.3. It encodes a protein that acts at the level of cell membranes during enamel mineralization. Its expression is more intense in the maturation stage than in the secretory stage during dental development.63 Its mode of transmission is autosomal recessive, accompanied by a hypomaturation AI. The dentine shows a normal thickness but with premature wear and yellow-brown color. A delay in dental eruption and even short stature has been described in patients suffering from mutation of the WDR72 gene.63,64
4. STIM1
The STIM1 gene (OMIM 605921, stromal interaction molecule 1) consists of 12 exons and is located on chromosome 11, position 11p15.4. It encodes a transmembrane protein with calcium binding domains, located in the endoplasmic reticulum. This protein is a calcium sensor that allows the transfer of calcium ions from the endoplasmic reticulum to the cell membrane. The STIM1 protein mediates the store-operated calcium entry (SOCE), which is necessary for the normal functioning of ameloblasts. The expression of STIM1 has been highly detected in ameloblasts maturation, compared with ameloblasts secretion during dental formation.42,43
Homozygous mutations with loss of function of the STIM1 gene cause combined immunodeficiency (CID) by STIM1 deficiency, a form of CID due to a dysfunction of calcium release-activated channels (CRAC). Patients with these mutations show hypocalcified AI leading to dentin loss.65
5. GPR68
The GPR68 gene (OMIM 601404, G protein-coupled receptor 68) has 1 exon and is located on chromosome 14, position 14q32.11. It encodes a proton-sensitive protein containing seven transmembrane helices, with histidine residues responsible for pH detection on the extracellular surface.66 Parry et al67 determined that GPR68 is expressed in the ameloblasts during all the amelogenesis stages.
Two homozygotic mutations (c.667_668delAA; p.Lys223Glyfs* 113 and c.221T> C; p.Leu74Pro) in the only exon of the GPR8 gene produce a hypomaturation AI, characterized by an opaque enamel, anterior open bite, and a loss of dental substance in permanent teeth.68
Table 2 summarizes the genes involved in non-syndromic AI, while Figure 3 shows the functions of the proteins encoded by these genes during dental enamel formation.
Table 2 Genes involved in non-syndromic amelogenesis imperfecta
| Gene (OMIM) | Position | Number of exones Transmission | Encoded protein | AI Type/associated syndrome | |
|---|---|---|---|---|---|
| Hypoplastic amelogenesis imperfecta | AMELX (amelogenin) OMIM 300391 | Xp22.2 | 7 Linked to X | Amelogenin (enamel matrix protein) | Hypoplastic or hypocalcified |
| AMBN (ameloblastin) OMIM 601259 | 4q13.3 | 13-AR | Ameloblastin (enamel matrix protein) | Hypoplastic | |
| ENAM (enamelin) OMIM 606585 | 4q13.3 | 9-AD/AR | Enamelin (Enamel matrix protein) | Hypoplastic | |
| LAMB3 (laminin Beta-3) OMIM 150310 | 1q32.2 | 23-AD | Laminin Beta-3 | Hypoplastic | |
| LAMA3 (laminin alfa-3) OMIM 600805 | 18q11.2 | 75-AD | Laminin Alfa-3 | Hypoplastic | |
| ACPT (acid phosphatase, testicular) OMIM 606362 | 19q13.33 | 11-AR | Testicular acid phosphatase | Hypoplastic | |
| Hypocalcified amelogenesis imperfecta | FAM83H (family with sequence similarity 83, member H) OMIM 611927 | 8q.24.3 | 5-AD | Intracellular protein- Involved in ameloblastic differentiation | Hypocalcified |
| C4ORF26 (chromosome 4 open reading frame 26) OMIM 614829 | 4q21.1 | 2-AR | Extracellular matrix acid-phosphoprotein | Hypocalcified | |
| SLC24A4 (solute carrier 24 A4) OMIM 609840 | 14q32.12 | 17-AR | Ion Exchanger | Hypocalcified | |
| ITGB6 (integrin Β-6 chain) OMIM 147558 | 2q24.2 | 15-AR | Integrins of epithelial cells | Hypocalcified | |
| AMTN (amelotin) OMIM 610912 | 4q13.3 | 9-AD | Amelogenin (Enamel matrix protein) | Hypocalcified | |
| Hypomaturation amelogenesis imperfecta | MMP20 (enamelysin) OMIM 604629 | 11q22.2 | 10-AR | Metalloproteinase | Hypomaturation |
| KLK4 (kallikrein 4) OMIM 603767 | 19q13.41 | 6-AR | Serine protease | Hypomaturation | |
| WDR72 (protein 72 with repeated WD) OMIM 613214 | 15q21.3 | 20-AR | Cytoplasmic protein - Enamel mineralization | Hypomaturation | |
| STIM1 (stromal interaction molecule 1) OMIM 605921 | 11p15.4 | 12-AR | Transmembrane protein-calcium sensor | Hypomaturation | |
| GPR68 (G protein-coupled receptor 68) OMIM 601404 | 14q32.11 | 1-AR | Enamel matrix pH sensor | Hypomaturation |
AD: autosomal dominant; AR: autosomal recessive. Source: Simancas V, Natera A, Acosta MG
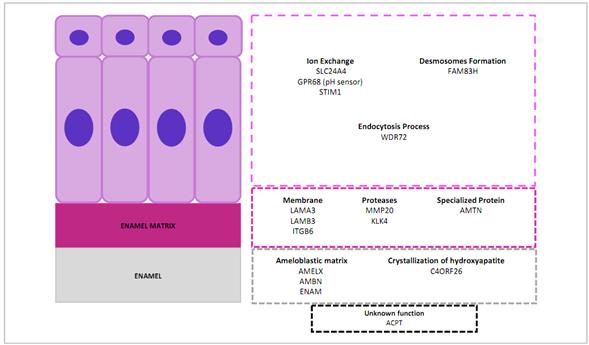
The enamel organ with ameloblastic cell area (purple), enamel matrix (violet), and enamel (grey). In the lower area, genes responsible for amelogenesis imperfecta with no known dental function at present (black box). Source: Simancas V, Natera A, Acosta MG.
Figure 3 Function of the proteins encoded by genes involved in amelogenesis imperfecta
CONCLUSIONS AND PERSPECTIVES
This literature review showed that mutations in 16 genes (AMELX, AMBN, ENAM, LAMB3, LAMA3, ACPT, FAM83H, C4ORF26, SLC24A4, ITGB6, AMTN, MMP20, KLK4, WDR72, STIM1, GPR68) are responsible for non-syndromic hypoplastic, hypocalcified, or hypomaturation AI.
Future research with a translational approach will allow the identification of new mutations or genes, enabling an evolution in the way of classifying and diagnosing the different types of AI. This new knowledge will certainly result in improved patient care and the development of new therapeutic options aimed at mitigating or eliminating the impact of AI as a source of social, aesthetic, and functional problems.

