










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkVitae
Print version ISSN 0121-4004
Vitae vol.15 no.1 Medellín Jan./June 2008
FARMACOLOGÍA Y TOXICOLOGÍA
DESARROLLO Y VALIDACIÓN DE UNA METODOLOGÍA ANALÍTICA POR ESPECTROFOTOMETRÍA ULTRAVIOLETA PARA LA DETERMINACIÓN DE COCAÍNA EN UN POLÍMERO SINTÉTICO
DEVELOPMENT AND VALIDATION OF AN ANALYTICAL METHODOLOGY FOR COCAINE HYDROCHLORIDE DETERMINATION IN A SYNTHETIC POLYMER BY ULTRAVIOLET SPECTROMETRY
Dorelly A. BONILLA Q.1; Luisa F. PEÑUELA B.1; Noralba SIERRA M. 1; Josías E. DÍAZ F.2; Jaime H. ROJAS B. 1*
1 Sección de Analítica, Departamento de Farmacia, Universidad Nacional de Colombia, A.A. 14490, Bogotá, Colombia
2 Laboratorio de Química, Fiscalía General de la Nación, Bogotá, Colombia
RESUMEN
Se presentan en este artículo los resultados del desarrollo y validación de una metodología analítica para la determinación de clorhidrato de cocaína en un polímero de látex sintético. El procedimiento consiste en la extracción de la cocaína con ácido sulfúrico 0,5 N y medida de la absorción a 233 nm. El método desarrollado es selectivo, lineal y preciso. La linealidad se obtiene entre 4 y 20 ppm, con límites de detección y de cuantificación de 1,6 y 4,0 ppm, respectivamente. La robustez de la metodología es igualmente estudiada, así como la estabilidad de la cocaína en ácido sulfúrico 0,5 N, requisito para el almacenamiento de muestras y estándares. Estas características, junto con su sencillez y exactitud, permiten que el método sea de fácil aplicación y adecuado para el objetivo propuesto. El método validado se aplica para la determinación del clorhidrato de cocaína en un polímero incautado por las autoridades legales.
Palabras Clave: Cocaina, polímero, cromatografía líquida, C18, detección ultravioleta, validación.
ABSTRACT
An ultraviolet spectrometry method was developed for the quantitative assay of cocaine hydrochloride in a latex plastic polymer. The absorption was determined in sulfuric acid 0,5N at 233 nm, after substance extraction using the same acid. Selectivity, linearity between 4 and 20 ppm and good reproducibility were shown by this method. Detection and quantification limits were 1,6 and 4,0 ppm, respectively. Method robustness was also verified, as well as cocaine hydrochloride stability in sulfuric acid 0,5N, medium where the samples and reference standards were stored. These properties, besides its simplicity and accuracy, make it suitable for achieving proposed objectives. The validated method was applied to the determination of cocaine hydrochloride in polymer confiscated by the law.
Keywords: Cocaine, polymer, reverse high performance liquid chromatography, C18, UV detection validation.
INTRODUCCIÓN
En las últimas décadas se ha visto aumentada la producción, distribución y comercialización de sustancias psicotrópicas, tales como la cocaína, cuyo consumo ilícito está sometido a fiscalización internacional. Este aumento se relaciona con la rápida diversificación en la obtención y distribución de este alcaloide, así como con su demanda, principalmente en los países industrializados. Para la pronta y rápida administración de justicia, las autoridades deben, además de ser competentes, disponer de metodologías analíticas validadas, confiables y de rápida ejecución. Entre las varias formas de camuflaje de estas sustancias ilegales, una que se ha vuelto común es su incorporación en algunos materiales tales como polímeros de naturaleza plástica. En Colombia es corriente el empleo de látex natural polimerizado para tal fin. Antes de proceder a la extracción y análisis cuantitativo de la cocaína presente en el polímero, se debe proceder a ensayos previos como pruebas de campo y cromatografía en capa delgada del polímero incautado, pruebas realizadas en el presente trabajo. Las muestras incautadas en Colombia frecuentemente se encuentran adulteradas y presentan una pureza entre 80 y 90% (1).
La cocaína, éster derivado del tropano, sufre reacciones de hidrólisis que dependen de la temperatura y del pH, presentando su máxima estabilidad a un pH cercano a 3,0 (2). Dentro de las diferentes técnicas que existen para la determinación de cocaína se encuentra la espectrofotometría ultravioleta en medio ácido a 233 y a 275 nm (3), la absorción al infrarrojo (4) y la cromatografía gaseosa con detector de ionización de llama, acoplada o no a espectrometría de masas (4 - 9, 10, 11), técnica que ha sido utilizada para la determinación de cocaína y de sus metabolitos en muestras de saliva (5). Por otra parte, la cromatografía líquida de alta eficiencia se ha empleado para su determinación en plasma (12), plasma y orina (13) y otras matrices (14). La literatura reporta igualmente la determinación de cocaína y otras drogas ilícitas por cromatografía en capa delgada de alta eficiencia (15) y por electroforesis capilar (16).
La revisión de la literatura no indica que se haya realizado la determinación de cocaína en una matriz constituida por el polímero derivado del látex o caucho natural. El caucho se obtiene del árbol por un tratamiento sistemático de sangrado, el cual consiste en hacer un corte en forma de ángulo a través de la corteza profundizando hasta el cambium (17). El látex contiene entre 30 y 36 % del hidrocarburo del caucho (isopreno), 0,3 - 0,7 % de cenizas, 1 - 2% de proteínas, 2% de resina y 0,5% de quebrachitol.
Para la validación de la metodología se siguieron las pautas y recomendaciones publicadas por la FDA (18), USP (19) y otros autores (20). Para el estudio de la exactitud de la metodología fue necesario sintetizar el polímero en el laboratorio, para lo cual se utilizó como monómero el estireno, como iniciador el peróxido de hidrógeno y como estabilizadores de la emulsión, Tween 20 y lauril sulfato de sodio. En el proceso de síntesis fue necesario optimizar el sistema de mezcla y agitación, la temperatura y el tiempo de reacción.
DESARROLLO EXPERIMENTAL
Equipos
Espectrofotómetro Unicam UV-VIS Tipo UV2-200, espectrofotómetro IR ATI Mattson Modelo Genesis Series FTIR, potenciómetro Mettler Toledo MA 235 pH/Ion Analyser, balanza analítica Mettler Toledo AB204-S, cromátografo de gases acoplado a masas Agilent Series 6890.
Reactivos
Además de los patrones secundarios de cocaína y de clorhidrato de cocaína, se emplearon los siguientes reactivos, todos de grado analítico: permanganato de potasio, ácido clorhídrico, ácido sulfúrico, carbonato de potasio, etanol, metanol, ácido acético glacial, ácido nítrico, hidróxido de sodio, biftalato de potasio, ácido perclórico, cloroformo, amoníaco, sodio metálico, tetracloruro de carbono, diclorometano, tetrahidrofurano, tolueno, ciclohexanona, acetato de etilo, acetato de amilo, cloruro férrico, cloruro de calcio, sulfato ferroso, nitrato de plata, piridina, molibdato de amonio y acetona.
Desarrollo de la metodología
El proceso de desarrollo experimental se realizó en tres etapas básicas: caracterización del polímero de incautación, estandarización de la técnica de extracción y la validación de la metodología analítica. Antes de proceder a la última etapa, se verificó la idoneidad del sistema analítico. El material volumétrico y el instrumental empleado se calibraron debidamente. Para el material volumétrico se siguió la Norma Internacional ISO 4787 y para la balanza, las recomendaciones de la Norma Técnica Colombiana NTC 2031 y la Organización Internacional de Metrología Legal (OIML). La exactitud de la longitud de onda del espectrofotómetro se verificó de acuerdo a la recomendaciones de la farmacopea británica (21). La longitud de onda para la determinación de la cocaína en ácido sulfúrico 0,1N se estableció en 233 nm, longitud a la cual se obtiene mayor sensibilidad, respecto de 275 nm, segundo máximo de absorción encontrado. La idoneidad del sistema se verificó al inicio, hacia la mitad y al final de la validación.
Con este propósito, se determinó la repetibilidad de la absorbancia (error fotométrico), la relación de absorbancias a longitudes de onda seleccionadas de acuerdo al espectro de absorción de la cocaína y la exactitud de la longitud de onda y de la absorbancia empleando una solución de dicromato de potasio (21).
Caracterización del polímero de incautación
Para la caracterización del polímero, identificación y posible composición, se realizaron las siguientes pruebas (22-24): análisis elemental luego de fusión sódica, reacción con piridina, ensayo de Liebermannn-Storch-Morawski y ensayos de solubilidad, así como análisis termogravimétrico (TGA), calorimetría de barrido diferencial (DSC) y espectroscopia infrarroja de reflectancia, pruebas éstas específicas para la caracterización de polímeros.
Estabilidad de las soluciones de cocaína
Algunas soluciones de cocaína pueden alterarse por crecimiento de hongos, aparición de color, fermentación y formación de productos de degradación, alteraciones favorecidas, entre otros, por factores tales como pH y temperatura. La estabilidad de una solución de cocaína de concentración 300 ppm se verificó por cromatografía gaseosa acoplada a masas usando tetracosano como estándar interno. Tres soluciones se almacenaron a temperatura ambiente y tres a cero grados centígrados.
Validación de la metodología analítica
Para la validación de la metodología analítica se procedió a la evaluación de los siguientes parámetros: linealidad, exactitud, precisión como repetibilidad y como precisión intermedia, concentración mínima detectable y concentración mínima cuantificable. Además, se verificó la robustez de la metodología, siguiendo las recomendaciones de Youden y Steiner (25).
Para esta etapa del estudio fue necesario elaborar una matriz a base de látex natural copolimerizada con estireno, utilizando como iniciador peróxido de hidrógeno y empleando Tween 20 y laurilsulfato de sodio como tensioactivos, matriz que fue enriquecida con una solución acuosa de clorhidrato de cocaína. Para fines del muestreo, la matriz obtenida se redujo a un tamaño de partícula de aproximadamente 1 mm, todo con el fin de mejorar la superficie de contacto con el solvente de extracción de la cocaína.
La selectividad, teniendo en cuenta los objetivos cualitativos y cuantitativos del trabajo, se comprobó frente a posibles contaminantes, agregados con fines de adulteración de la cocaína, tales como lidocaína, procaína, fenacetina, efedrina y cafeína (26). Estas sustancias pueden ser identificadas por espectrofotometria y por ensayos preliminares. Igualmente con el propósito de verificar la calidad de la muestra, se realizaron ensayos a la gota con reactivo de Tanred y Scott y cromatografía en capa delgada con tres sistemas solventes diferentes (4). En este caso se utilizaron cromatoplacas de sílica gel 60 F254, como revelador el reactivo de Dragendorff y como sistemas de desarrollo ciclohexano-tolueno-dietilamina (75:14:10), cloroformo-metanol-amoníaco (90:10:1) y metanol-butanol-ácido clorhídrico (60:40:2,8).
RESULTADOS Y DISCUSIÓN
Caracterización del polímero de incautación
El análisis elemental demostró ausencia de halógenos, azufre y fósforo, por lo cual la búsqueda se orientó hacia un polímero orgánico, exceptuando caucho vulcanizado con azufre o una resina de caseína (ausencia de fósforo). Las pruebas de solubilidad confirman la anterior conclusión.
Los análisis térmicos y calorimétricos en el polímero de incautación indican señales que corresponden a la cocaína, a sus productos de descomposición y al plastificante empleado.
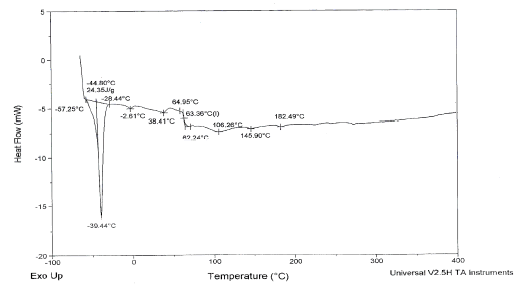
Como se observa en la gráfica de calorimetría diferencial de barrido, (DSC) obtenida con el polímero incautado luego de la extracción de la cocaína con ácido sulfúrico 0,5N (Figura1), aparece una señal a -39.44°C con un valor de Cp de 24.35J/g, pero ninguna señal en la región de 190°C, en adelante, indicativa de la presencia de cocaína en el polímero como tal. La señal a -39.44°C corresponde al plastificante empleado en su elaboración con el propósito de conferirle propiedades de elasticidad.
Figura 1. Gráfica de DSC de la matriz del polímero incautado
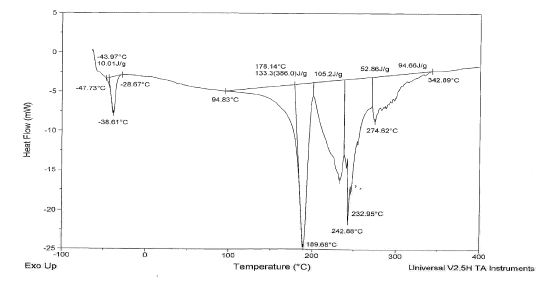
En el polímero incautado (Figura 2), se observa además una señal, no encontrada en la Figura 1, que corresponde al punto de fusión de la cocaína (189.68°C), y que indica claramente la presencia de la droga. Las señales obtenidas a 242,88, 232,95 y 274,62°C, se deben a las temperaturas de degradación de la cocaína. Se reportan tres temperaturas diferentes, pues la cocaína puede presentar diferentes tamaños de cristales, resultantes de las formas de cristalización del alcaloide producto de su inclusión en el polímero. En este caso, la señal por el plastificante utilizado presenta un valor de Cp que desciende a 10.01J/g, debido posiblemente a una interferencia de la cocaína en la cantidad de calor absorbido por el material en este intervalo de temperatura. Las señales adicionales que se presentan a diferentes temperaturas como inflexiones en la Figura 1, demuestran que el polímero de incautación se encuentra en el grupo de las poliolefinas y cauchos. La temperatura de transición vítrea del látex no se alcanza a observar, pues correspondería a -70°C, valor por fuera de la curva.
Figura 2. Gráfica de DSC del polímero incautado
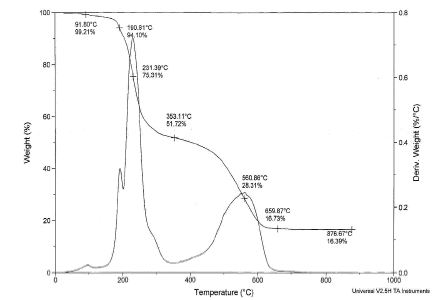
En el ensayo de caracterización por análisis térmico diferencial (TGA) del polímero incautado (Figura 3), los valores indicados sobre la curva decreciente corresponden a la pérdida de peso de la muestra por el incremento en el valor de la temperatura, con caídas que representan la volatilización de las sustancias presentes. El punto de inflexión a 190,81°C, con una pérdida de peso reducida a 94,10%, es cercano al punto de fusión de la cocaína y al inicio de su volatilización. Una mejor visualización se tiene en la curva de primera derivada. Sobre la figura, la curva sobre la que se observan picos, corresponde a la primera derivada y permite una mejor visualización. La señal a 231,39°C correspondería a la degradación y posterior volatilización, así como a la presencia de humedad o de alguna otra sustancia. La respuesta para la primera derivada a 560.86°C corresponde al inicio de la degradación del polímero.
Figura 3. Gráfica de TGA para el polímero incautado
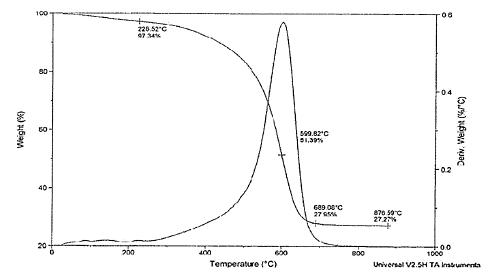
En la Figura 4, obtenida con el polímero incautado libre de cocaína, no se obtienen señales por debajo de 500°C, lo cual evidencia la ausencia de cocaína. Se obtienen, en cambio, señales entre 599,82°C y 689,08°C, temperaturas de inicio de degradación del polímero. Esta curva demuestra que la pérdida de componentes volátiles del polímero sin cocaína es constante hasta que empieza su degradación.
Figura 4. Gráfica de TGA para la matriz del polímero incautado
Estandarización del método de extracción
La eficiencia de la extracción de la cocaína con ácido sulfúrico 0,1N se evaluó a tiempos entre 0,5 y 6,0 horas, no observándose cambios significativos en el proceso de extracción a partir de las 2,0 horas. Posteriormente se analizó el proceso de extracción frente a diferentes concentraciones de ácido sulfúrico. No presentaron diferencias en el porcentaje de extracción al utilizar concentraciones 0,1 y
0.5 N de ácido sulfúrico. Como medio de extracción se estableció ácido sulfúrico 0,5 N.
Estabilidad de las soluciones de cocaína
La cocaína (benzoilmetilecgonina), por su naturaleza de éster, sufre reacciones de hidrólisis, las cuales son promovidas y catalizadas por un medio ácido, medio establecido para el proceso de extracción eficiente de la droga. La estabilidad, durante 26 días, se determinó mediante cromatografía gaseosa – espectrometría de masas utilizando tetracosano como estándar interno, para seis diferentes soluciones de cocaína, con concentraciones entre 200 y 320 ppm. De estas soluciones, tres se mantuvieron a la temperatura del laboratorio y las restantes a 0°C. Los resultados demostraron descomposición significativa de la cocaína al tercer día de iniciado el estudio para las soluciones mantenidas a la temperatura del laboratorio y al quinto día para las soluciones almacenadas a 0°C. En consecuencia se recomienda, en caso de ser necesario, mantener refrigeradas las soluciones patrón de cocaína y analizarlas antes del tercer día de preparadas o, en el caso de muestras, de ser sometidas al proceso de extracción.
Validación de la metodología analítica
Selectividad. La selectividad de la metodología, analizada frente a las sustancias utilizadas comúnmente para adulterar la cocaína (efedrina, cafeína, procaína, fenacetina y lidocaína), se estableció mediante la determinación de los espectros de absorción entre 200 y 350 nm. Los espectros indican absorción variable para cada una de ellas a 233 nm, longitud de onda empleada para la cuantificación de la cocaína. En la Tabla 1 se reportan los valores de absorbancia a 233 nm de los adulterantes mencionados.
Tabla 1. Absorbancia de algunos adulterantes a 233 nanómetros
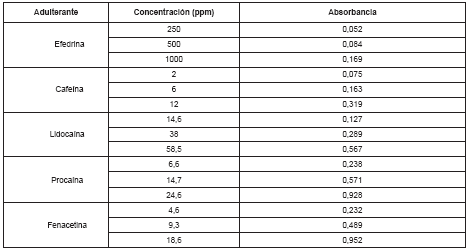
De acuerdo a los resultados obtenidos, la absorción encontrada causaría una interferencia en la cuantificación de la cocaína por el método espectrofotométrico, si estas sustancias llegaren a estar presentes. Por ello, se hace necesario una identificación previa de la muestra por pruebas de campo o por cromatografía en capa delgada. En la Tabla 2 se presentan los resultados obtenidos con los tres sistemas solventes mencionados anteriormente.
Tabla 2. Valores de Rf para algunos adulterantes en tres sistemas cromatográficos
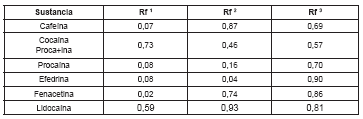
1 ciclohexano-tolueno-dietilamina (75:14:10)
2 cloroformo-metanol-amoníaco (90:10:1)
3 metanol-butanol-ácido clorhídrico (60:40:2,8)
Los espectros de absorción ultravioleta de los componentes de la matriz, látex natural, estireno, Tween 20, laurilsulfato de sodio y peróxido de hidrógeno a las concentraciones de ensayo, no presentaron prácticamente absorción a 233 nm, demostrándose de esta manera la selectividad de la metodología frente a estos compuestos.
Linealidad. La proporcionalidad de la respuesta, para el sistema y para el método, se verificó con concentraciones de cocaína entre 4 y 20 ppm, tres réplicas para cada nivel de concentración. Para el método, la curva de calibración, ajustada por mínimos cuadrados, se representa en la Figura 5.
Figura 5. Curva de calibración para la linealidad del método
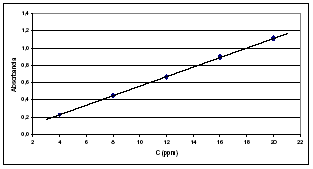
En la Tabla 3 se reportan los valores experimentales para intercepto y pendiente, así como los valores de t para la prueba t de Student, con trece grados de libertad y una probabilidad de error del 5%. En la tabla se encuentran igualmente los límites calculados de acuerdo al error estándar de cada uno de ellos. Los valores experimentales de t indican convergencia al origen (t experimental < t de la tabla) y un valor de pendiente significativamente diferente de cero (t experimental > t de la tabla), tanto para el sistema como para el método. Se observa que el cero se ubica dentro de los límites calculados para los interceptos.
Tabla 3. Resultados del test de Student para intercepto y pendiente*
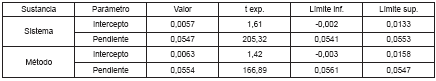
*t de la tabla (gl: 13, a: 0,05): 2,16
Los valores del coeficiente de determinación para sistema y método fueron respectivamente 0,9997 y 09998. Los valores experimentales de F de la prueba de Fisher para el análisis de varianza, tanto para el sistema como para el método, indican una regresión altamente significativa y un desvío de la linealidad no significativo.
Precisión. La precisión a nivel de repetibilidad, para sistema y método se determinó realizando 10 análisis independientes de tres réplicas de soluciones preparadas de concentración 8, 12 y 16 ppm. Los valores del coeficiente de variación obtenidos, en ningún caso superan el 2%, límite establecido como coeficiente de variación para metodologías analíticas (ICH, USP).
La precisión intermedia, sistema y método, se analizó de acuerdo a un diseño experimental consistente en el análisis de una solución de cocaína de concentración 8 ppm por parte de dos analistas, durante cuatro días consecutivos, tres réplicas por analista y tres lecturas para cada análisis. Los coeficientes de variación ponderados para cada uno de los dos analistas fueron para el sistema 1,99 y 1,60% respectivamente. Los valores para el método fueron 1,23 y 0,38%.
Exactitud. Para el estudio de la exactitud del método, la matriz de polímero sintetizada se enriqueció con tres niveles de concentración de cocaína, 4, 8 y 16%. Para cada nivel se realizaron tres extracciones independientes, alcanzándose un porcentaje de recuperación promedio de 83,58 %. De acuerdo al test t de Student, la recuperación hallada es significativamente diferente del 100%. Sin embargo, el valor puede ser considerado como aceptable si se tiene en cuenta la complejidad de la matriz de poliestireno.
Igualmente se determinó si la concentración influye de alguna manera en la variabilidad de los resultados. Para ello se utilizó el test de Cochran para comparación de varianzas. El valor de G experimental hallado fue 0,652, valor inferior a 0,898, correspondiente al obtenido de la tabla de G de Cochran para k igual a tres (niveles de concentración), para n igual a tres (réplicas para cada nivel de concentración) y una probabilidad de error de 0,05%. Como conclusión se establece que el nivel de concentración no influye en la variabilidad de los resultados y que las varianzas para los distintos niveles de concentración no son significativamente diferentes entre sí.
Límites de detección y de cuantificación. Para establecer los valores de la cantidad mínima detectable (CMD) y de la cantidad mínima cuantificable (CMC) se utilizó el método de la relación señal/ruido, método bastante utilizado cuando se trata de determinar estas cantidades. A 233 nm, como CMD se encontró un valor de 9,73 x 10-3 ppm y como CMC un valor de 2,95 x 10-2 ppm. Estos límites se verificaron experimentalmente, encontrándose que no podían diferenciarse de la magnitud del ruido. De acuerdo a ello, se procedió a determinar los límites siguiendo el método de las diluciones seriadas o método de la curva de calibración a bajas concentraciones. Se trabajaron los siguientes niveles de concentración: 0,4, 0,8, 1,2, 1,6 y 2,0 ppm, tres réplicas para cada nivel.
Los resultados, de acuerdo a la prueba t de Student, indican una pendiente significativamente de cero y un intercepto no significativamente diferente de cero. El cero de la curva de regresión se encuentra dentro de los límites calculados para el intercepto. Por otra parte, el análisis de varianza permite concluir que la regresión es significativa y un desvío de la linealidad no significativo.
Sin embargo, se observan elevados valores del coeficiente de variación, principalmente para las tres primeras concentraciones. Por esta razón se propone como CMD una concentración de 1,6 ppm, nivel al cual el coeficiente de variación hallado fue 2,27%, y como CMC una concentración de 4 ppm, primer punto de la curva de calibración del sistema.
Matrices del polímero se enriquecieron con concentraciones de 2 y 4 ppm. Los porcentajes de recuperación, promedio de tres réplicas, fueron 68,95 y 71,78% para 2 y 4 ppm, respectivamente.
Robustez. La robustez del método se estableció de acuerdo al diseño de Placket Burtman y realizando pequeñas variaciones en algunos parámetros del método (Tabla 4). Al comparar los valores de diferencia obtenidos para cada factor en estudio, se encontró que la diferencia media observada para cada nivel (superior e inferior) fue menor a la diferencia del producto de la desviación estándar de la precisión del método por la raíz cuadrada de 2 para el factor longitud de onda. Se concluye que el método es sólido frente a las pequeñas variaciones realizadas en la temperatura, tiempo de lectura, concentración de ácido sulfúrico y analista.
Tabla 4. Influencia de la variación de algunos parámetros en la respuesta
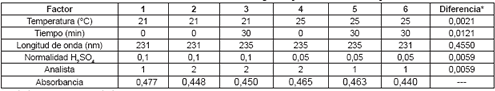
*media límite superior – media límite inferior
desviación estándar del métodox1.4142 = 0,0111
CONCLUSIONES
Se implementaron y validaron algunos parámetros de una metodología analítica para la cuantificación de cocaína en un polímero de látex natural por espectrofotometría ultravioleta, polímero utilizado por el narcotráfico como medio de camuflaje de la droga. De acuerdo a varios métodos de análisis de identificación realizados, se caracterizó la matriz en estudio como compuesta por un látex natural copolimerizado. Para la validación se hizo necesario sintetizar una matriz de látex copolimerizado con características similares, tanto en estructura como en apariencia, flexibilidad y resistencia física. Gracias al proceso de extracción estandarizado y al método de muestreo propuesto, se obtuvieron resultados que permitieron inferir sobre el contenido del analito presente en la totalidad de la muestra incautada. Durante la etapa de validación, la metodología demostró ser precisa, lineal, robusta y selectiva frente a los componentes de la matriz. Los límites de detección y de cuantificación encontrados fueron 1,6 y 4 ppm, respectivamente. La metodología es además sencilla en su realización sin incurrir en costos excesivos, permite la entrega rápida de resultados y la disminución de tiempo de trabajo de los peritos forenses, todo lo cual beneficia la pronta administración de justicia por parte de los organismos del estado encargados de ella. Es necesario proceder a ensayos previos por cromatografía en capa delgada para detectar sustancias que se utilizan a veces para rebajar y adulterar la cocaína. Si estas sustancias, como por ejemplo, lidocaína, procaína, fenacetina, llegaren a ser detectadas, se hará necesario determinar el contenido de cocaína por una metodología selectiva (HPLC).
REFERENCIAS BIBLIOGRÁFICAS
1. Barrios A, Giral LM. Validación de una metodología para el análisis de cocaína por espectrofotometría ultravioleta. , [Tesis de grado] Bogotá: Universidad de América; 2002. [ Links ]
2. Connors K. Curso de análisis farmacéutico. Ensayo del medicamento. Barcelona: Reverté; 1981. p. 371-376. [ Links ]
3. Clarke's. Isolation and identification of drugs, 2ª ed. Londres: The Pharmaceutical Press; 1986. p. 226-227. [ Links ]
4. UNDCP, Aplicación de técnicas cualitativas y cuantitativas para sustancias de uso indebido, Tercer curso básico nacional y primero internacional. Guadalajara de Buga: 1998. Módulo 1, Numerales 3.2 y 3.3. [ Links ]
5. Martínez M, Troncoso C. Validación de una metodología analítica para la determinación de cocaína y sus metabolitos a partir de muestras de saliva por cromatografía de gases. Bogotá: UNAL; 2000. [ Links ]
6. Castro HJ. La espectrometría de masas y la cocaína. Revista del Instituto Nacional de Medicina Legal y Ciencias Forenses 2004; 18: 46-52. [ Links ]
7. Pujadas M, Pichini S, Civit E, Santamariña E, Pérez K, De la Torre R. A simple and reliable procedure for the determination of psychoactive drugs in oral fluid by gas chromatography–mass spectrometry. J Pharm Biomed Anal. 2007; 44: 594-601. [ Links ]
8. Saito T, Mase H, Takeichi S, Inokuchi S. Rapid simultaneous determination of ephedrines, amphetamines, cocaine, cocaine metabolites, and opiates in human urine by GC–MS. J Pharm Biomed Anal. 2007; 43: 358-363. [ Links ]
9. Alvarez I, Bermejo AM, Tabernero MJ, Fernández P, López P. Determination of cocaine and cocaethylene in plasma by solid-phase microextraction and gas chromatography–mass spectrometry. J Chrom B. 2007; 845: 90-94. [ Links ]
10. Valente-Campos S, Yonamine M, de Moraes RL, Alves Silva O. Validation of a method to detect cocaine and its metabolites in nails by gas chromatography–mass spectrometry. Forensic Sci Int 2006; 159: 218-222. [ Links ]
11. Fucci N, de Giovanni N, Chiarotti M. Simultaneous detection of some drugs of abuse in saliva samples by SPME technique. Forensic Sci Int 2003; 134: 40-45. [ Links ]
12. Fernández P, Morales L, Vázquez C, Bermejo AM, Tabernero MJ. HPLC–DAD determination of opioids, cocaine and their metabolites in plasma. Forensic Sci Int. 2006; 161: 31-35. [ Links ]
13. Johansen SS, Bhatia HM. Quantitative analysis of cocaine and its metabolites in whole blood and urine by high-performance liquid chromatography coupled with tandem mass spectrometry. J Chrom B. 2007; 852: 338-344. [ Links ]
14. Wood M, Laloup M, del Mar Ramirez M, Jenkins KM, Young MS, Ramaker JG, De Boeck G, Samyn N. Quantitative analysis of multiple illicit drugs in preserved oral fluid by solid-phase extraction and liquid chromatography–tandem mass spectrometry. Forensic Sci Int. 2005; 150: 227-238. [ Links ]
15. Antonilli C, Suriano C, Grassi MC, Nencini P. Analysis of cocaethylene, benzoylecgonine and cocaine in human urine by high-performance thin-layer chromatography with ultraviolet detection: a comparison with high-performance liquid chromatography. J Chrom B Biomed Sci App. 2001; 751: 19-27. [ Links ]
16. Gottardo R, Bortolotti F, De Paoli G, Pascali J, Miksik I, Tagliaro F. Hair analysis for illicit drugs by using capillary zone electrophoresis-electrospray ionization-ion trap mass spectrometry. J Chrom A. 2007; 1159: 185-189. [ Links ]
17. http://www.textoscientificos.com/caucho/natural. Consultado: Octubre 15 de 2007. [ Links ]
18. Guidance for Industry, Bioanalytical Methods Validation, Draft Guidance, Food and Drug Administration, Centre for Drug Evaluation and Research CDER, Mayo de 2001. [ Links ]
19. The United States Pharmacopoeia, USP 30. The Pharmacopoeia Convention; 2007. p 588. [ Links ]
20. Quatrochi OA, Abelaira SI, Laba RF. Introducción a la HPLC, Teoría y Práctica. Buenos Aires; 1992. p. 324-327. [ Links ]
21. The British Pharmacopoeia BP 2004. British Phamacopea Comission Secretariat, Her Majestic Office, London, A213, pag. A141. [ Links ]
22. Billmeyer F. Texbook of polymer science. New York: 1962. p 25-26, 90-96, 105-106, 383-386. [ Links ]
23. Noriega RI. Guías de laboratorio de caracterización de polímeros. Bogotá: Universidad Nacional de Colombia; 1984. p 40-57. [ Links ]
24. Hummel DO. Atlas of polymer and plastics análisis, 2ª. Ed. Vol. 1, p 2006-2022. [ Links ]
25. Youden WJ. The Collaborative Test. J Assoc Anal Chem. 1990; 71: 195-197. [ Links ]
26. United Nations International Drug Control Programme, Informe 2000-2003. New York: ONU; 2004. [ Links ]
AGRADECIMIENTOS
Se agradece a la Fiscalía General de la Nación y a su Laboratorio de Química por el apoyo financiero para la realización del presente trabajo.
* Autor a quién debe dirigirse la correspondencia: jhrojasb@unal.edu.co

