











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Solar cells are devices for the production of environmental friendly renewable energy resources and the best candidates to replace highly polluting non-renewable energies, such as fossil fuels and nuclear energy [1]. One way to obtain sustainable energy through solar cells is the use of a panel which converts solar energy into electricity [2]. There is a new trend using the organic molecules as active layer components in solar cells [3-5].
The selection of material is crucial to build a solar cell relying on organic molecules as the active layer [6], because they must be suitable to be used as acceptor or electron donor to fulfill the energy requirements in order to generate the photovoltaic effect [7].
The donor-acceptor (D-A) mixture receives photons from the solar radiation that generates an excited state known as exciton, which is formed by a hole-electron pair [8-10]. The further steps for the electric current generation process are as follows: the flow of the excitons to the D-A interface; charge separation of excitons at the D-A interface and finally diffusion and collection of the carriers to electrodes [11]. This cycle is necessary to generate electric current. Each compound of the D-A mixture must have certain values in their physicochemical parameters to know if that compound is the best choice for the active layer. The active layer is one of the most important components of the organic solar cells (OSCs), because this electric current (electron flow per unit time) is generated by the interaction of this layer with sunlight. The function of the active layer is to perform as donor-acceptor (D-A) mixture [12, 13].
The Density Functional Theory (DFT) is the most used and accurate method for the determination of the physical and chemical properties of compounds that are investigated [14-17].
Yuksek et al. [18] reported that B3LYP functional is one of the most efficient functionals especially for 1H-NMR calculations. Siskos et al. [19] also used B3LYP functional with different basis sets for theoretical calculations of some keto-enol tautomer including their 1H-NMR and reported a good consistency with experimental results.
Xerri et al. [20] investigated the role of π-bond characteristics in dyes for the application in solar cells through the DFT method. The π-bonds are directly related to the most important physicochemical properties. For example, the highest occupied molecular orbital (HOMO) has the ability to donate the electrons [21]. In addition, its energy corresponds to the ionization potential. On the other hand, the lowest unoccupied molecular orbital (LUMO) has the ability of accepting electrons, and its energy corresponds to the electron affinity [21, 22].
Hachmann et al. [23] calculated theoretical properties of several organic compounds which have potential use in organic photovoltaics.
In this work two groups of organic molecules, synthesized by Zhen et al. [24], and Dang et al. [25], were analyzed by DFT calculations, and the experimental values were compared to the theoretical results via linear regression analysis.
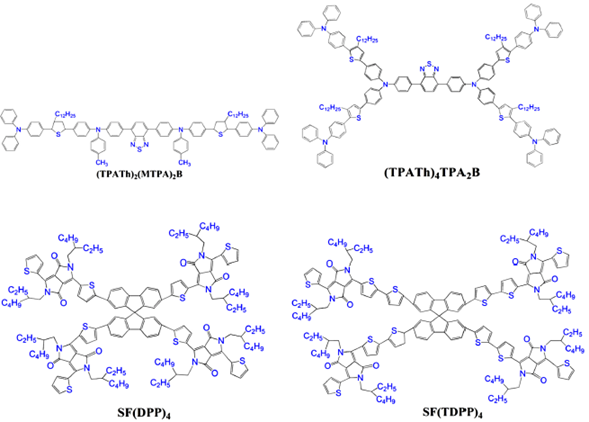
The structures of the molecules studied are presented in figure 1. The (TPATh)2(MTPA)2B and (TPATh)4TPA2B molecules were synthesized and characterized [24, 25]. Both systems contain triphenylamine which is considered as a promising unit for the transport of charges, light emitting and photovoltaic material, due to its good donation of electrons and high capacities of hole transport [24]. On the other hand, SF(DPP)4 y SF(TDPP)4 molecules were synthesized and characterized by Dang et al., both molecules show adequate experimental levels of energy of the highest occupied molecular orbital (HOMO) and the lower unoccupied molecular orbital (LUMO), a great capacity of electron transfer [25].
These four molecules were considered for the theoretical study because they have a wide absorption in UV-Visible region, acceptable levels of their energy bands and are economical to synthesize and no any theoretical properties are available so far. A broad and strong absorption in the UV-Vis region of the electromagnetic spectrum is fundamental in a molecule, whose purpose is to be used in a solar cell, since if the absorption occurs in a short range of wavelength, it cannot be used properly [24,25].
Computational details
Gaussian 09 package [26] was used to determine physicochemical properties of the molecules above-mentioned. The input files were prepared via using the visualization program, GaussView 5.0 [27]. The molecules investigated have been computed and analyzed with the restricted Density Functional Theory (DFT) employing Becke's three parameter hybrid exchange functional with Lee-Yang-Parr correlation functional (B3LYP) [28] with the 6-31G(d) basis set. Tetramethylsilane (TMS) was used as reference for the theoretical 1H NMR calculations of (TPATh)2(MTPA)2B molecule using GIAO [29] method.
Additionally, the frontier molecular orbitals (FMOs) of all the optimized structures were obtained with the same basis and analyzed as well. Once the energy of FMOs is obtained (ionization energy, I=-EHOMO, and electron affinity, A=-ELUMO), it is possible to calculate the other physicochemical properties such as global hardness (η) and softness (α), electronic chemical potential (µ), and the global electrophilicity index (ω) [22]. The properties mentioned are calculated [30] according to the equations given in Table 1.
TABLA 1 Ionization energy (I), electron affinity (A), global hardness (η), softness (σ), electronic chemical potential (µ), and the global electrophilicity index (ω). Total energy and comparison of the experimental and theoretical HOMO-LUMO gap energies (H - L gap). All the energy values are given in eV.
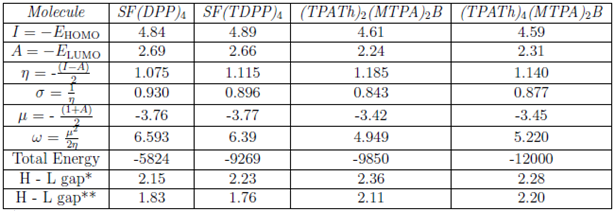
*Theoretical energy gap
**Experimental energy gap
Results and discussions
The total minimum energy values of the optimized structures for each compound based on their conformations studied are given in Table 1. As it is well-known, the geometry optimization of a system is the first step to calculate the other physicochemical parameters [31].
The view of the FMOs upon the structures optimizations of the four molecules analyzed is given in figure 2a, figure 2b, figure 2c and figure 2d.
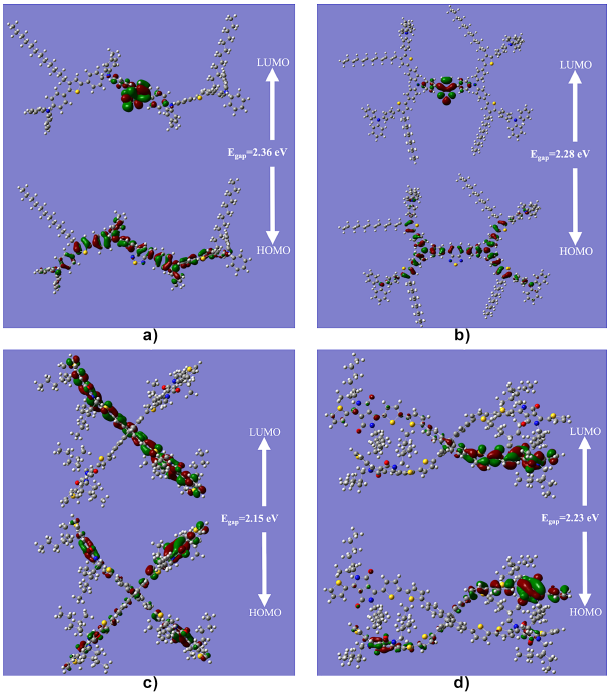
Figura 2 The optimized structures of a) (TPATh) 2 (MTPA) 2 B, b) (TPATh) 4 TPA 2 B, c) SF(DPP) 4 and d) SF(TDPP) 4 molecules.
Analyzing the total minimum energy of triphenylamine based molecules obtained after the optimization the structures proposed by the authors [24, 25], the molecule labelled as (TPATh)2(MTPA)2B (-253715 eV) has less energy than the (TPATh)4TPA2B (-159429 eV). On the other hand, the energy values for spirobifluerene based molecules show that SF(TDPP)4 (-328165 eV) has less energy comparing to SF(DPP)4 (-268083 eV). The optimization process was carried out over the structures presented in figure 1 without any further conformational space analysis.
Once the optimization of the organic molecules is done, it is possible to obtain several parameters necessary to know if they present the conditions to be used in the active layer of a solar cell, one of many important parameters are the energies HOMO y LUMO. The parameters mentioned, methods and basis set which have been used to oftriphenylethylamine, and diketo-pyrrolo-pyrrole (DPP) are the most common and accurate ones for these type of calculations [32, 33]. Martinez, Zarate et al. [34] used a similar method to calculate theoretical properties of pentacene and the benzo[g]benz[6,7]indeno[1,2-b]fluorene, which would act as components of OSCs.
Zhan et al. [35] used the DFT method to calculate molecular orbital energies and determine the theoretical properties of some inorganic and organic compounds. The authors have reported a good consistency between theoretical properties such that ionization energy (I), electron affinity (A), global hardness (h) among the others with the experimental data [35]. The authors have compared some experimental data that they obtained with the theoretical. They also used the effect of the larger basis sets on the accuracy of the calculations. One of the molecules that the authors have calculated its ionization energy is 1,3-butadiene, which has a conjugated system and can be correlated to the systems that we have analyzed. Both experimental and theoretical ionization energy values for that compound has been reported as 8.972 and 9.082 eV, respectively. The ionization energy for the compounds in this study is between 4.5-4.9 eV. It is quite reasonable that different compounds would have different energy values depending on their structures, the theoretical method, and the basis set used. As the compounds analyzed in this study do not have any experimental data about ionization energy, hardness, softness, electron affinity, we could not specify those properties based on the theoretical values obtained.
On the other hand, Oshi et al. [36] have reported some anthracene and tetracene derivatives'theoretical properties. Even though the compounds we analyzed contain some heteroatoms like nitrogen and sulfur and much bigger than anthracenes and tetracene, due to the aromaticity we may compare the theoretical values such as ionization energies, electron affinities, and energy gaps. The ionization energies, electron affinities, and energy gaps values (in eV) reported by the authors are for anthracene, and its derivatives are in the range of 0.5-4.6, 7-10.8 and 0.25-3.6, respectively. The same values (in eV) for tetracene and its derivatives have been reported in the range of 1-4.9, 6.5-10, and 0.4-2.8. The values stated in this study are in the range indicated by Oshi et al. [36]. So, as seen even though we have used different functional and basis sets since compounds analyzed contain aromatic groups, the theoretical values obtained are similar to the literature.
The energies and energy gap of frontier molecular orbitals for (TPATh)2(MTPA)2B, (TPATh)4TPA2B, SF(DPP)44 and SF(TDPP)4 calculated are shown in the figure 2 (a,b,c,d) and Table 1.
The LUMO isosurfaces of (TPATh)2(MTPA)2B and (TPATh) 4TPA2B molecules are found to be a part of the π-conjugated system and forming nodes (see figure 2a and figure 2b). Specifically, in the whole central block composed of the electrophile benzothiadiazole group takes the role to provide greater electron affinity according to Kutkan et al. [4], and slightly in the two phenyl groups adjacent as shown in figure 1.
For the case of the HOMO in the aforementioned molecules, the electrons in p orbitals form the n-conjugated system, concentrating in the benzene ring of the central block of benzothiadiazole in the nucleophilic arms of triphenylamine [5, 37] next to the central block. The π-conjugated system occurs especially at the locations where thiophene and triphenylamine groups are located [38].
Regarding the SF(DPP)4 molecule, the LUMO is found in the section of the π-conjugate system forming nodes throughout the two arms (see figure 2c) [39]. In case of SF(TDPP)4 (see figure 2d), the LUMO is localized only on one arm. This is probably due to the first structure drawn to create the input file [40]. The structures of HOMO for SF(DPP)4 and SF(TDPP)4 molecules show the interaction between the electrons in p orbitals forming n bonds (see figure 2c and figure 2d).
As seen in figure 2c and figure 2d HOMOs are delocalized especially on the diketopyrrolopyrrole and in the benzene ring. These results are in accordance with the paper published by Sambathkumar et al. [41].
Once the LUMO and HOMO energies of each molecule have been determined, it is possible to calculate the LUMO-HOMO energy gap (band energy or bandgap) which is used to evaluate the molecule for different purposes [10,42].
The error percentages between experimental and calculated energy gap values are -17.48, -26.7, -11.84, -3.63 for SF(DPP)4, SF(TDPP)4, (TPATh)2 (MTPA)2 B, and (TPATh)4 TPA2 B molecules, respectively. The results show that the experimental and theoretical energy gap values for both (TPATh) 2 (MTPA) 2 B, and (TPATh)4TPA2B molecules are relatively have a good fit. In the case of SF(DPP) 4, and SF(TDPP) 4 molecules the error is between 17-27%.
B3LYP functional was used for 1H-NMR calculations, since it gives accurate theoretical results [18, 19]. To get more insight on the molecular structure of the studied systems, the 1 H-NMR chemical shifts were computed. Figure 3 shows the linear regression analysis between the experimental and calculated 1 H-NMR chemical shifts for (TPATh)2 (MTPA)2B molecule. The R2 value is too close to 1 which means that the experimental and calculated 1 H-NMR values are very similar and the characterization of the small molecule by experimental 1 H-NMR technique is supported by this high value of R2.
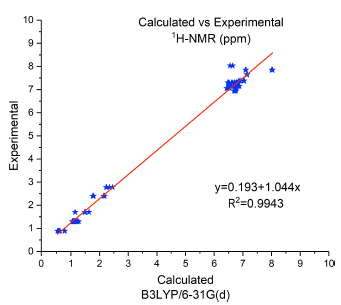
FIGURA 3 Comparison of experimental and calculated 1 H-NMR chemical shifts for (TPATh) 2 (MTPA) 2 B molecule.
The comparison of experimental and theoretical 1 H-NMR chemical shifts was done to confirm the reliability of the experimental results given in the paper published [24].
Conclusions
In this study two different series of small molecules used in organic solar cells were analyzed theoretically by DFT (using B3LYP/6-31G(d)) method. The energy gap values calculated for SF(DPP)4 and SF(TDPP)4 are very close to experimental values confirming the experimental study. On the other hand, the values calculated for (TPATh)2(MTPA)2B and (TPATh)4(MTPA)2B are relatively different than the experimental.
The similarity between the experimental and theoretical 1H-NMR values for (TPATh)2(MTPA)2B molecule can be accepted as a support to the experimental characterization.
The calculated energy gap, 1 H-NMR chemical shifts and corresponding linear regression analysis confirm the potential use of the molecules synthesized [24,25] as donor on organic solar cells.

