










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de Ingeniería
Print version ISSN 0121-4993
rev.ing. no.33 Bogotá Jan./June 2011
HyRPNI Algorithm and an Application to Bioinformatics
El algoritmo HyRPNI y una aplicación en bioinformática
Gloria Inés Alvarez V
PhD. en Reconocimiento de formas e inteligencia. Profesora asociada, Pontificia Universidad Javeriana. Cali, Colombia. galvarez@javerianacali.edu.co
Jorge Hernán Victoria M
Ingeniero de Sistemas, Pontificia Universidad Javeriana. Cali, Colombia. jhvictoria@javerianacali.edu.co
Enrique Bravo M
PhD. en Ciencias-Biología. Profesor titular, Universidad del Valle. Cali, Colombia. enrique.bravo@correounivalle.edu.co
Pedro García G
Doctor en Ciencias de la Computación, Departamento de Sistemas Informáticos y Computación, Universidad Politécnica de Valencia. Valencia, España. pgarcia@dsic.upv.es
Recibido 21 de abril de 2009, modificado 14 de enero de 2011, aprobado 21 de junio de 2011.
PALABRAS CLAVES
Aprendizaje automático, bioinformática, lenguajes formales.
RESUMEN
Proponemos un algoritmo de inferencia gramatical para lenguajes regulares que permite ahorrar cómputo al usar dos criterios diferentes para elegir los estados a ser procesados, un criterio se usa en la primera fase del proceso de inferencia (al principio) y el otro en el resto del proceso. Realizamos experimentos para observar el desempeño del algoritmo, para aprender sobre el tamaño ideal de su primera fase y para mostrar su aplicación en la solución de un problema específico en bioinformática: la predicción de sitios de corte en poliproteínas codificadas por virus de la familia Potyviridae.
KEY WORDS
Bioinformatics, formal languages, machine learning.
ABSTRACT
We propose a grammar inference algorithm for regular languages which saves computational cost by using two different criteria to choose states to be processed: one in the first phase of the inference process (the beginning) and another for the rest of the process. We applied experiments to observe performance of the algorithm, to learn about the best size of its first phase and to show results of its application to solve a specific problem in Bioinformatics: the cleavage site prediction problem in polyproteins encoded by viruses of the Potyviridae family.
INTRODUCTION
Grammar inference is a technique of inductive learning, that belongs to the syntactic approach of machine learning. We develop grammar inference algorithms based on the merge of states in the mood of well-known algorithms like RPNI [7] or red-blue [5]. Our goal is to contribute to developing new algorithms which are less expensive in computational cost and prone to be applied to real world problems.
We base this research on the idea that the first merges in the inference process are the most important [2]. Therefore it is acceptable to spend more computational resources making these decisions as soon as possible with the available information. Since final merges are not so determinant in the quality of the final model, they can be chosen in a simpler and cheaper way. An appropriate balance of both kinds of decisions would yield an effective and efficient inference algorithm.
The rest of the paper is structured as follows: Section 2 presents some useful definitions and notation conventions. Section 3 describes our proposed algorithm. Section 4 presents the cleavage site prediction problem, a bioinformatics problem considered to be solved by the algorithm. Section 5 presents some results obtained in the three experiments with both synthetic and real data. Finally, our conclusions and future work are presented.
PRELIMINARIES
Let Σ be a finite alphabet and Σ* the free monoid1 generated by Σ with the concatenation as the internal operation and ε as the neutral element. A language L over Σ is a subset of Σ*. Let ω be a word, that is, ω ∈ Σ* and the length of w is denoted as |ω|.
The transition function δ is defined in symbols, but it can be extended to words as follows: Let q ∈ Q, s ∈Σ* if | s | > 1 that is, s = s1, . .., sn then δ (q, s) = δ (δ(q, s1, . .., sn-1), sn). A word x is accepted by A if δ (q0, x) ∈F. The set of accepted words by A is denoted by L (A), and is called the language of A.
A word x is accepted by M if Φ(x) = 1. The set of accepted words by M is denoted by L(M).
ALGORITHM HYRPNI
Hy-RPNI (Hybrid Regular Positive and Negative Inference) is the name of our grammar inference proposal. It consists of two phases: in the first phase, a heuristic based on a score is used to choose the pairs of states to be merged; the second phase chooses pairs of states for merging in lexicographical order (Definition 2.1). Algorithm 1shows the general strategy for inference process. It receives as input a set of positive (D+) and negative (D-) samples, and the size of the first phase (phaseOneSize), measured as a number of merges; and returns the smallest DFA (Definition 2.2) consistent with training samples.
List S defined in line 2 stores the states that will be in the final hypothesis. List T defined in line 3 stores the states in the frontier of S (Definition 2.7). In line 4, S is initialised with q0, the initial state of the AMPAT M (Definition 2.6). The function calculateFrontier (lines 5 and 17) assigns to list T the frontier of S. The counter countPhase is defined in line 6 starting from zero, it increases each time a merging is done and it allows the end of the first phase to be detected. While statement starts in line 7 and stops when T becomes empty and the final hypothesis is obtained. Line 9 verifies the current phase of the algorithm, and applies the corresponding method: heuristic or lexicographical. In both cases, only if a merge is done will the countPhase be incremented by one. Algorithm returns the DFA associated to the final Moore Machine (Definitions 2.3 and 2.4).

Merging of states must preserve the consistency of the machine with respect to training samples. In our approach, two states can be merged if they are undistinguished (Definition 2.8). The merging functions of the algorithm first determine if states can be merged and proceed to merge them when possible. The function HeuristicMerge explores all the pairs of state candidates to be merged, formed by a state from set S and one from frontier T. Each pair able to be merged is scored to reflect the degree of support, in the training sample, to the belief that this merge will be correct, which is to say that it will lead us to a right model for the target language. The Function LexicographicalMerge merges the first pair of states, one from set S and the other from set T, in lexicographical order. In both alternatives if a state from T cannot be merged with any state from S, this state is promoted to set S, which is to say that the state is inserted in set S and deleted from set T.
PROPERTIES
HyRPNI is a deterministic, polynomial algorithm whose time complexity is upper bounded by the expression O(kn4 + (m- k)n3) where k is a constant representing the size of the first phase in terms of number of merges done, m is the total number of merges done and n is the size of the initial AMPAT, which is to say the number of states of the initial model. This complexity expression has two terms corresponding to the inference phases respectively: kn4 corresponds to the cost of applying HeuristicMerge algorithm (with complexity O(n3)) k times into the main cycle of line 6 in the Hy-RPNI algorithm. The second term: (m - k)n3 corresponds to the cost of performing the rest of merges with a cost of n2 each one into the main cycle in line 6 of the Hy-RPNI algorithm. The worst case of this algorithm happens when k = m and all the merges are done in the first phase. In this case, the complexity is the same as the Red-blue algorithm and computes scores during the whole inference process. The best case happens when k = 0 and the complexity is the same as the RPNI algorithm. Convergence of this algorithm is provided because it has been proved that the convergence of a state merging inference algorithm is obtained independently of the order in which merges are done [8] when we are inferring DFAs as in this case. Notice that changing the criteria to merge states only establishes a different order of merging.
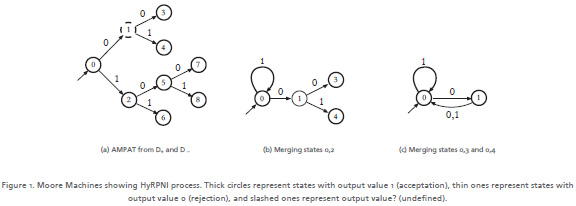
EXAMPLE
Consider the AMPAT shown in Figure 1a, when HyRPNI is used, it starts with the first phase, that applies a heuristic method, S and T lists are set as S = {0} and T = {1, 2}. At this point the heuristic function assigns two scores, one for the pair of states (0, 1), and the other for (0, 2). Using the heuristic function, scores are (0, 1) = 199 and (0, 2) = 499. This means we should merge states 0 and 2. After the merge shown in the Figure 1b, the first phase ends and the process continues in lexicographical order. The next merges are states 0, 3 and 0, 4, in that order. The inferred automaton using Hy-RPNI is shown in the Figure 1c.
BIOINFORMATIC PROBLEM
Cleavage site prediction problem consists of predicting the position in a sequence of amino acids where a particular subsequence with a specific meaning or function begins (or ends). This problem is present in any genome, from viruses to human beings. We are interested in cleavage site prediction on potyviruses, because they are pathogenic for many important crop plants such as beans, soybean, sugarcane, tobacco and others with economic and alimentary impact in our region. The prediction of cleavage sites can make the understanding and control of the diseases caused by these kind of viruses easier.
The potyviral genome is expressed through the translation of a polyprotein which is then cut by virusencoded proteinases at specific sites in the sequence of amino acids, resulting in 10 functionally mature proteins responsible for the infection and virus replication. The functions of these viral-encoded proteins are partially understood. Prediction of cleavage sites is not trivial because even though there are patterns of symbols that mark these places, these patterns are quite variable. Because of the complexity of the cleavage site sequences, the use of algorithms makes the detection of specific features of those points easier. The prediction of cleavage sites allows isolating specific segments to be studied and facilitates the analysis and annotation of the data obtained experimentally as well as allowing for their comparison with those existing in databases, such as GenBank.
There are different programs approaching the cleavage site prediction problem, in several contexts and particular species. Some of them arise from previous work and tend to improve the methods and performance of the predecessors. SigCleave from the Emboss repository predicts the site of cleavage between a signal sequence and the mature exported protein. Its predictive accuracy is estimated to be around 75- 80% for both prokaryotic and eukaryotic proteins [9]. Another program available is PeptideCutter which predicts potential cleavage sites cleaved by proteases or chemicals in a given protein. NetChop produces neural network predictions for cleavage sites of the human proteasome [10]. ChloroP predicts the chloroplast transit peptides (cTP). ChloroP can distinguish between cTPs and other proteins with a correlation of 0.76, and it can locate the cleavage site within three residues from annotated position in about 60% of the cTPs [6]. The PSORT program is an integrated system of several prediction methods, using both sorting signals and global features[3], which has subsequently grown to an entire family of prokaryotic and eukaryotic protein localisation predictors. PSORT II predicts on eukaryotic sequences, establishing their sub cellular localisation from two classes: endoplasmic reticulum, extracellular and Golgi are merged into one category of secretory proteins, whereas the rest: cytoplasmic, mitochondrial, nuclear, peroxisomal and vacoular, are merged in to a single non-secretory category [4]. A successor of ChloroP is TargetP, which provides predictions of cTPs, mTPs and secretory signal peptides (SPs). For the prediction of cleavage sites SP, TargetP uses the SP cleavage site prediction from SignalP. SignalP is one of the most used methods (it uses Hidden Markov Models and Artificial Neural Networks). It predicts the presence of signal peptidase to cleavage sites. SignalP produces both classification and cleavage site assignment, while most of the other methods classify proteins as secretory or non-secretory [1]. As well as being the most used it is also the most compared method. SignalP outperforms PSORT-II in signal peptide discrimination [1] and outperforms TargetP in the discrimination between secretory and non-secretory proteins. In spite of the availability of these and other software tools to predict cleavage sites, it should be noted that none of them is designed to segment polyproteins of Potyviridae family viruses. Thus, none of them could be used to solve the problem described at the begin 49 ning of this section. In fact, as far as we know, this is the first paper about the prediction of cleavage sites over the Potyviridae family, so there is no background information on which to make any comparisons. None of these tools use grammatical inference (GI). Instead most of them use hidden Markov Models and/or neural networks. Two main reasons encourage us to apply GI: it does not require changes in data representation nor does it require assumptions about past dependencies among symbols in the sequence as traditional methods do.
EXPERIMENTAL RESULTS
We present three experiments where we tested the HyRPNI algorithm. The first one shows the behavior of the recognition rate as the size of first phase grows on a well-known corpora of synthetic data. The second experiment explores the variation in the best value for the length of the first phase of the algorithm in a new synthetic corpora. Finally, the third experiment applies the HyRPNI algorithm to the cleavage site prediction problem on polyproteins from genomes of Potyviridae family.
The first experiment is defined on synthetic data, using a corpus available in the academic community designed by Denis et.al.[3]. We used 30 target automata with sizes between 2 and 22 states. Thirty automata were trained varying the size of the first phase from 1 to 10 and considering samples of maximum length 20 (group A), 30 (group B) and 40 (group C). We measured recognition rate (on a test set of one thousand samples different from the training ones) and execution time; we report the average over the thirty languages learnt. Results are presented in Table 1. Notice that five merges in the first phase of the algorithm are enough to reach best performance. We have compared this behaviour with other well-known algorithms: RPNI and red-blue and our results are comparable with those obtained by Red-blue when heuristic selection is achieved during all the inference process.
The second experiment was conducted on a new corpus of synthetic data which we generated that includes larger target automata. We generated target automata of 5, 10, 20, 50 and 100 states (30 automata from each size). For each size thirty automata were generated and used to label samples. The goal of this experiment is to better understand the behaviour of the parameter corresponding to the size of the first phase of the algorithm. Training samples were 100 in all the cases while test samples were 1000 and were disjoint of training ones. The total length of the inference processes was established in thirty five merges on average. We reported the average over the thirty languages learnt for recognition rate and execution time. The second experiment allows us to observe the behaviour of HyRPNI algorithm while learning larger languages. We are interested in the size of the first phase which allows reaching the highest recognition rate with respect to the total number of merges in the inference process. We consider samples of maximum length 20 (group A), 30 (group B) and 40 (group C).
Table 2 presents results of the second experiment. We observe that recognition rates diminish when the size of target automata increases, this may be explained by the fact that all the trainings were done with 100 samples. For a small target automata it is enough information, but for a larger one, 100 samples are very few and don't yield high recognition rates. We noticed that the best values for the size of the first phase vary without following a steady trend. For example, In the results for group A, for target automata of size 10 we achieved the best performance with 14 merges in the first phase, but if the size of the target automata increases to 20 the best value decreases to 6. However, if the size of target automata increases again to 100 states, the best value increases to 23, which is more than half of the inference process because we know the complete inference process has nearly 35 merges. In group C, we get smaller best sizes for the first phase, but with sudden variations. We compared these recognition rates against those obtained by red-blue algorithm, which applies heuristic merge during all the inference process and they are comparable in all the cases. We notice that even when we need a long first phase to achieve our best recognition rates there is a savings on computations of HyRPNI with respect to red-blue (comparison are not reported due to space limitations).We concluded from this experiment that although HyRPNI allows saving some computational costs compared with other algorithms with similar recognition rates, the magnitude of the savings varies from one case to another. In consequence, the size of the first phase of the algorithm should be tuned experimentally according to the specific task.
The third experiment applied HyRPNI to the problem of predicting cleavage sites in sequences from polyproteins encoded by the genome of viruses of the Potyviridae family. Our purpose is to learn (to create?) a model for recognising each of the nine cleavage sites present in the coding portion of the viral genome once it is translated. The training samples were obtained from sequences published in www.dpvweb.net/potycleavage/index.html which contains approximately 50 samples for each cleavage site. We are using all the available labelled sequences we know from plant virus databases. As HyRPNI needs negative samples, we used positive samples of other sites as negative samples for each given model. We trained with cross validation of four blocks. The amino acid sequence is considered one window at a time. Three lengths of window were explored: in the first case we suppose the cleavage site is located between the fourth and fifth symbol. For this reason we refer to this window as 4/1. In a similar way, we experimented with windows 14/1 and 10/10. Three values were tried on for the first phase size parameter: 5, 10 and 25 merges. We selected these values considering the results of former experiments and estimations of the total number of merges in the inference process. We quantified the number of samples in each one of the following categories: tp (true positive, positive test samples well classified by the automaton), tn (true negative, negative test samples well classified), fp (false positive, negative test samples classified as positives) and fn (false negative,positive test samples classified as negatives). From these values for a given test set, we can calculate several measures:
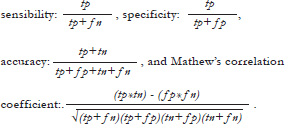
Table 3 shows the results obtained for the first cleavage site called P1-HCPro due to the names of the segments it separates. Each cleavage site has its own peculiarities for recognition, but due to space limitations we are reporting only one segment. Results in each position of the table are the average over four executions of cross validation. It can be noticed that the execution time is around 30 seconds with window 4/1, but increases to several minutes with 14/1 and 10/10 windows. In spite of this, all experiments take an acceptable time. Regarding accuracy values, window 4/1 has the best values, beyond 98 percent. Observing the balance between sensitivity and specificity, window 4/1 has the best performance again. Among the parameters of HyRPNI algorithm, in window 4/1, first phase of length 10 has a better correlation coefficient. Following these data, we could conclude that the model to recognize the first cleavage site should be trained using 10 merges in the first phase of the algorithm and using a window 4/1. The same kind of analyses have been applied to model other cleavage sites, giving us clues about the best values for parameters of the method.
CONCLUSIONS
We present a new grammar inference algorithm designed to get high recognition rates because it carefully selects states to be merged in the first part of the inference process and it saves temporal cost because it changes the criterion of selection to a computationally less expensive one in the second phase. We show three experiments, two on synthetic data and one on a real problem to illustrate the possibilities of the method. From the first experiment it follows that the first phase (expensive phase) could be short maintaining high recognition rates. The second experiment shows that as the size of the target language grows the ideal size of the first phase changes in an unpredictable way. Nevertheless, it shows that in all the cases it is possible to save computational cost and keep high recognition rates. Finally, the third experiment illustrates the application of HyRPNI to predict cleavage sites. We obtained significant high recognition rates. In future work we will apply these models to build an application to do automatic segmentation of polyproteins coming from Potyviridae family genomes and to determine whether GI overperforms other traditional methods.
NOTAS AL PIE
1. The free monoid is a set that has a binary operation over it, such that the result of applying the operation to two elements of the set produces another element in the set; also, the binary operation needs to be associative and the set needs to have a neutral element.
REFERENCES
[1] J. Dyrlov Bendtsen, H. Nielsen, G.von Heijne and S. Brunak. "Improved Prediction of Signal Peptides: SignalP 3.0". Journal in Molecular Biology JMB. Vol. 340, 2004, pp. 783-795. [ Links ]
[2] O. Cichello and S. C. Kremer. "Beyond EDSM". Lecture Notes in Artificial Intelligence. Vol. 2484, 2002, pp. 37-48. [ Links ]
[3] F. Denis, A. Lemay and A. Terlutte. "Learning Regular Languages Using RFSAs". Theoretical Computer Science. Vol. 313, 2004, pp. 267-294. [ Links ]
[4] O. Emanuelsson, S. Brunak, G. von Heijne and H. Nielsen. "Locating Proteins in the Cell Using TargetP, SignalP and Related Tools". Nature Protocols. Vol. 2, No. 4, 2007, pp. 953-971. [ Links ]
[5] K. J. Lang. "Random DFA's can be Approximately Learned from Sparse Uniform Examples". Proceedings of the Fifth Annual ACM Workshop on Computational Learning Theor y, 1992, pp. 45-52. [ Links ]
[6] H. Nielsen, S. Brunak and G. von Heijne. "Machine Learning Approaches for the Prediction of Signal Peptides and Other Protein Sorting Signals". Protein Engineering. Vol. 12, No. 1, 1999, pp. 3-9. [ Links ]
[7] J. Oncina and P. García. "Inferring Regular Languages in Polinomial Update Time". Pattern Recognition and Image Analysis, 1992, pp. 49-61. [ Links ]
[8] G. Alvarez. Estudio de la Mezcla de Estados Determinista y No Determinista en el Diseño de Algoritmos para Inferencia Gramatical de Lenguajes Regulares. Universidad Politécnica de Valencia, 2007. [ Links ]
[9] Emboss SigCleave. Fecha de consulta: diciembre 2010. Disponible: http://emboss.bioinformatics.nl/cgibin/emboss/help/sigcleave [ Links ]
[10] NetChop 3.1. Fecha de consulta: diciembre 2010. Disponible: http://www.cbs.dtu.dk/services/NetChop/ [ Links ]

