










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Med
Print version ISSN 0121-5256On-line version ISSN 1909-7700
rev.fac.med vol.15 no.2 Bogotá July/dic. 2007
ARTÍCULO
EL ADIPOCITO COMO ÓRGANO ENDOCRINO. IMPLICACIONES FISIOPATOLÓGICAS Y TERAPÉUTICAS ADIPOSE TISSUE AS AN ENDOCRINE ORGAN. PATHOPHYSIOLOGICAL AND THERAPEUTIC IMPLICATIONS MARITZA PÉREZ MAYORGA M.D. Endocrinólogaa* a Centro de Investigaciones Facultad de Medicina, Universidad Militar Nueva Granada. * Correspondencia: maperezm@umng.edu.co. Dirección postal: Universidad Militar Nueva Granada, Facultad de Medicina, Centro de Investigaciones.Tr. 3 N° 49-00. Bogotá, D.C. Colombia. El concepto de tejido adiposo como el sitio de almacenamiento del exceso de energía ha cambiado en la última década y en la actualidad éste se reconoce como un tejido con alta actividad metabólica y como un órgano endocrino importante, capaz de establecer comunicación con el resto del cuerpo mediante la síntesis y la liberación de moléculas activas llamadas adipoquinas, las cuales actúan localmente y a distancia por medio de efectos autocrinos, paracrinos y endocrinos. También están implicadas en el control de la ingesta de alimentos, en el balance de energía, en el peso corporal, en la homeostasis de glucosa, en el metabolismo lipídico, en la angiogénesis, en la fibrinólisis, en los procesos pro y antinflamatorios, en el desarrollo sexual y en la reproducción. Todas estas sustancias tienen un papel clave en la patogénesis del síndrome metabólico, integrado por un grupo de factores de riesgo de origen metabólico que se acompañan a su vez de un riesgo aumentado de diabetes mellitus tipo 2 y de enfermedades cardiovasculares, de prevalencia cada vez mayor en el mundo y considerados actualmente como una pandemia. Una mejor comprensión de la función endocrina del tejido adiposo y de su papel en la patogénesis de estas enfermedades conducirá al desarrollo de una terapéutica más racional para estos desórdenes. Este artículo pretende integrar el conocimiento acerca de la fisiología del tejido adiposo con su papel en las patologías asociadas y con las posibles opciones terapéuticas para su prevención y tratamiento. Palabras clave: tejido adiposo, citoquinas, inflamación, resistencia a la insulina. Abstract Key words: adipose tissue, cytokines, inflammation, insulin resistance. Introducción La consideración del tejido adiposo como reservorio de energía ha venido cambiando con los años y desde 1987 se lo consideró como el principal sitio de producción de esteroides sexuales (1). Posteriormente, en 1994, se identificó la leptina, una hormona producida principalmente en el adipocito, que condujo al establecimiento del tejido adiposo como órgano endocrino (2). Es un tejido que no está conformado solamente por adipocitos maduros, sino también por fibroblastos, células endoteliales y macrófagos, los cuales constituyen cerca del 50% de su contenido celular total. Todas son células altamente activas y funcionan como una unidad integrada. El tejido adiposo consiste, funcionalmente, en dos tipos de tejido diferente: el pardo y el blanco. Los adipocitos pardos se especializan en la producción de calor a partir de su almacenamiento lipídico y se encuentran únicamente en mamíferos. Difirieren de los adipocitos blancos en la expresión de la proteína desacoplante 1 (UCP-1), que disipa como calor en forma de ATP, el gradiente de protones generado por la cadena de transporte de electrones en la membrana mitocondrial. Morfológicamente los adipocitos pardos son multiloculares, contienen menos lípidos que los blancos, siendo particularmente ricos en mitocondrias. En humanos, el tejido adiposo pardo rodea el corazón y los grandes vasos durante la infancia, tendiendo a desaparecer con el tiempo, de forma tal que sólo escasas células se pueden encontrar en los cojinetes grasos (3). Por otra parte, el tejido adiposo blanco, por medio de la captación de ácidos grasos libres, de su conversión en triacilgliceroles y de su hidrólisis a ácidos grasos libres como fuente energética, provee una reserva de combustible a largo plazo. En los mamíferos representa la principal reserva de energía y se distribuye en múltiples depósitos corporales, tanto interna como subcutáneamente, al igual que en nódulos linfáticos y en músculo esquelético. Adicional al almacenamiento de combustible, el tejido adiposo blanco puede actuar como un aislante térmico y como protector de órganos. A medida que la prevalencia del sobrepeso y de la obesidad se incrementan, también lo hace el interés por un mejor conocimiento del tejido adiposo, de tal forma que está demostrada la importancia de este tejido en patologías que se derivan de su exceso y de su deficiencia, las cuales se asocian con insulino resistencia, con hiperglucemia, con dislipidemia, con hipertensión y con estados protrombóticos y proinflamatorios, componentes todos del síndrome metabólico, el cual se define como un grupo de factores de riesgo de origen metabólico que se acompañan de un riesgo aumentado de diabetes tipo 2 y de enfermedades cardiovasculares. Este síndrome está alcanzando proporciones epidémicas, calculándose que cerca de una cuarta parte de la población mundial tiene esta patología (3,4) Para explicar la asociación entre adiposidad y patología se han postulado tres teorías (4): la portal/visceral, la lipodistrofia adquirida y la teoría del paradigma endocrino. La primera le otorga un papel central al aumento de grasa visceral y a su drenaje directo a la circulación porta, que lleva a una inhibición de la acción de la insulina, disminuyendo la oxidación de la glucosa y su utilización muscular, aumentando la producción hepática de glucosa y de lipoproteínas de muy baja densidad, además de un efecto lipotóxico sobre la célula beta, eventos todos que podrían explicar la relación entre obesidad, insulino resistencia y diabetes de tipo 2. La teoría de la lipodistrofia adquirida (o síndrome de almacenamiento ectópico de grasa) se basa, primero, en la presencia de insulino resistencia severa y de diabetes, probablemente como consecuencia del almacenamiento de lípidos en el hígado, en el músculo y en las células beta pancreáticas, en pacientes con lipodistrofia; segundo, en la correlación entre insulino resistencia y el grado de infiltración lipídica en el tejido muscular esquelético, hígado y probablemente en las células beta en los pacientes obesos y, tercero, en el hecho de que el incremento en el tamaño de la célula grasa se asocia con insulina resistencia y diabetes, representando esta situación una incapacidad para expandirse y para acomodar un alto flujo de energía (5). Por último, la teoría del paradigma endrocrino se basa en el conocimiento del tejido adiposo como un órgano endocrino que produce péptidos bioactivos, que no sólo influencian al adipocito en una forma autocrina y paracrina, sino que afecta varias funciones metabólicas a distancia (4) y en la presencia, en este tejido, de numerosos receptores que le permiten responder a diversas señales aferentes desde varios sistemas hormonales y el sistema nervioso central. Gracias a estos conocimientos, el impacto del tejido adiposo en el metabolismo es cada vez más evidente, habiéndose descrito su papel en la regulación de la masa grasa, en la homeostasis de nutrientes, en la respuesta inmune, en el control de la presión arterial, la homeostasis, la masa ósea y en las funciones reproductiva y tiroidea (6). Por todo esto y destacando el aporte del adipocito a la fisiopatología de algunas enfermedades, esta revisión se centrará en la descripción de las principales funciones endocrinas descritas para el tejido adiposo blanco y en su impacto en las diversas patologías, así como en la descripción de las intervenciones terapéuticas en uso y en las potencialmente útiles para tratar las patologías asociadas. Producción adipocitaria endocrina Las adipoquinas, diversas en cuanto a su estructura proteica y a su función fisiológica, establecen una red de comunicaciones con tejidos y órganos como el músculo esquelético, la corteza adrenal, el cerebro, el sistema nervioso simpático (4). Incluyen citoquinas clásicas, factores de crecimiento y quimitotácticos, factores del complemento y proteínas comprometidas en la regulación de la presión arterial, la homeostasis vascular, el metabolismo lipídico, la homeostasis de glucosa, la angiogénesis y la osteogénesis (Tabla 1). Leptina: Descubierta en 1994 como producto del gen Ob en el ratón genéticamente obeso (Ob/Ob), la leptina fue la primera adipoquina propuesta como moduladora de adiposidad y a la fecha es la mejor estudiada (7,8,9,10). Del griego leptos que significa delgado, es un polipéptido de 16 κ que contiene 167 aminoácidos, con homología estructural a las citoquinas. Es secretada por los adipocitos en proporción directa a la masa de tejido adiposo, al contenido de triglicéridos y al estado nutricional, siendo mayor en el tejido subcutáneo que en el tejido visceral y disminuyéndose su producción con el ayuno prolongado (11). La expresión y secreción de leptina se incrementa por la insulina, por los glucocorticoides, por el factor de necrosis tumoral alfa (TNF-α), por los estrógenos y por proteínas alfa de unión al activador CCAAT (enhancer-binding protein alfa). De igual forma se disminuye por la actividad adrenérgica β3, por los andrógenos, por los ácidos grasos libres, por la hormona de crecimiento y por los agonistas del receptor nuclear PPARγ (Peroxisome Proliferator-Activated Receptor Gamma). Circula en el plasma parcialmente unido a proteínas plasmáticas, ingresando por difusión al sistema nervioso central, por medio de uniones capilares en la eminencia media y por un transporte saturable a través de receptor en el plexo coroide (4). Sus receptores, miembros de la superfamilia de receptores de citoquina clase I, se expresan en el sistema nervioso central y periférico, identificándose variantes de ellos, aunque al parecer es la forma primaria del mismo, la que media la mayoría de sus efectos (12). Las isoformas ObRa y ObRc son receptores cortos que se localizan predominantemente en la barrera hematoencefálica, en donde parecen desempeñar un papel transportador, sobre todo el ObRa. La forma ObRb, estructuralmente más larga, contiene un dominio de señalización intracelular para la vía JAK/STAT (Janus activated kinase/signal transducer activator of transcription), abunda en el hipotálamo y se asume como el principal efector de las acciones de esta hormona. Por último, la isoforma ObRe es la fracción soluble, la cual transporta la leptina por el torrente circulatorio, regulando su depuración y su vida media (13,14). Este receptor se expresa en bajos niveles en numerosos tejidos, pero en el hipotálamo medio basal, particularmente, en el núcleo arcuato, en el núcleo ventromedial y en el dorsomedial, se encuentra en altos niveles (15,16). En el núcleo ventromedial del hipotálamo la leptina estimula los receptores citoquin quinasa 2 (CK2), la síntesis de la hormona estimulante de los melanocitos y el transcriptor regulado por cocaína-anfetamina (CART), moléculas, que por vía paracrina, estimulan los receptores 3 y 4 de melanocortina del núcleo lateral provocando saciedad. A ese mismo nivel inhibe péptidos orexígenos, como el neuropéptido Y y la proteína relacionada con el gen-agouti. La leptina inhibe la lipogénesis y estimula la lipólisis, reduciendo los niveles de lípidos intracelulares en el músculo esquelético, el hígado y las células beta pancreáticas, mejorando de esta manera la sensibilidad a la insulina. En el músculo, esta sensibilidad se logra mediante la inhibición de la malonil Co-A, lo que incrementa el transporte de ácidos grasos a la mitocondria para la beta oxidación. En el sistema límbico estimula la recaptación de dopamina, bloqueando así el placer de comer y a través del núcleo cerúleo, activa el sistema nervioso simpático que conlleva un incremento del gasto energético en reposo (17). Se podría afirmar entonces que la principal acción de la leptina es la de actuar como señal de saciedad. Durante el ayuno la leptina desciende rápidamente y estimula también la liberación de glucocorticoides, la reducción de tiroxina, de las hormonas sexuales y de la hormona del crecimiento. Por su acción angiogénica puede afectar la estructura vascular y por medio del receptor plaquetario de leptina contribuye a la trombosis arterial. In vitro estimula la producción de intermediarios reactivos del oxígeno como resultado de la activación de macrófagos (18). Adiponectina: Esta hormona ha sido identificada por diferentes grupos y ha recibido diferentes nombres, entre ellos el de proteína de adipocito relacionada con complemento (Acrp30, por sus siglas en inglés) por su homología con el factor C1q del mismo. Se trata de una proteína 30κ, codificada por el gen APM1 (3q27) compuesto por tres exones codificantes, expresada casi que exclusivamente en el tejido adiposo blanco. Puede sufrir modificaciones postraduccionales de hidroxilación y glucosilación, produciendo trímeros, hexámeros, o isoformas de alto peso molecular. Posee un dominio amino terminal similar a colágeno, un dominio carboxiterminal globular que media multimerización y una estructura terciaria que guarda alta similitud con el factor de necrosis tumoral alfa, a pesar de no tenerla en su estructura primaria (19). Para la adiponectina se han propuesto dos tipos de receptores: uno, con dos proteínas transmembranales similares y con homología a receptores de unión a proteína G conocidos como adipoR1 y adipoR2 y otro, la T-cadherina muscular, expresada primariamente en el músculo y que funciona como receptor de alta afinidad para la adiponectina globular. AdipoR2 se expresa principalmente en el hígado y funciona como un receptor de afinidad intermedia para la adiponectina de alto peso molecular, habiéndose reportado una isoforma de alto peso molecular que promueve la actividad de la adenosin monofosfato protein quinasa (AMPK) en los hepatocitos. De otra parte, sólo los trímeros activan AMPK en el músculo mientras que los hexámeros y la isoforma de alto peso molecular activan el factor nuclear kappa beta (NF-κβ). Estas actividades divergentes se podrían explicar por diferencias en el patrón de expresión específica para cada tejido (4) de forma tal que los efectos biológicos de la adiponectina dependen no sólo de sus concentraciones circulantes y de las propiedades de las diferentes isoformas, sino también de la expresión tejido específico de los subtipos de su receptor (20). Es interesante el hecho de que la adiponectina circula en concentraciones extraordinariamente altas, representando cerca del 0,01% de toda la proteína plasmática (21) y contrario a las otras adipoquinas, sus niveles tienen correlación inversa con la masa corporal, con la insulino resistencia y con los estados inflamatorios (22,23). La interleuquina 6 (IL-6) y el TNF-α son potentes inhibidores de su expresión y de su secreción (14). La adiponectina inhibe la adhesión de monocitos a las células endoteliales, la transformación de macrófagos en células espumosas y la activación de células endoteliales. Ejerce también un efecto sinérgico con la leptina para mejorar la sensibilidad a la insulina (24). Dentro de sus efectos metabólicos se encuentran: la mejoría de la insulino sensibilidad a nivel hepático, el descenso del flujo de ácidos grasos libres no esterificados, el incremento de la oxidación de grasa y la reducción de la salida de glucosa hepática. En el músculo, la adiponectina estimula el uso de glucosa y la oxidación de los ácidos grasos (25). Factor de necrosis tumoral alfa (TNF-α): El TNF-α fue la primera proteína secretada por el tejido adiposo y por las células estromovasculares a la que se le descubrió un papel en la homeostasis de glucosa (26). Es una proteína transmembranal, clivada en una proteína biológicamente activa de 17κ, que se expresa mayormente en el tejido subcutáneo comparado con el tejido visceral y que ejerce sus acciones vía receptores de tipo I y II, expresados por el adipocito (27). El ARN mensajero (mARN) del TNF-α del tejido adiposo se correlaciona con el índice de masa corporal, con el porcentaje de grasa corporal y con la hiperinsulinemia. Su efecto endocrino directo parece menos importante que los efectos indirectos, que resultan de la modulación auto y paracrina de los ácidos grasos no esterificados que disminuyen su captación por el tejido adiposo, así como la inhibición en la expresión de adiponectina y de interleuquina 6 (28). En el tejido adiposo, el TNF-α reprime genes comprometidos en la captación de ácidos grasos no esterificados y de glucosa, suprime genes para factores de transcripción comprometidos en adipogénesis y lipogénesis, e interviene en la expresión de varios factores secretados por los adipocitos, incluyendo adiponectina e interleuquina 6 (29). Interleuquina 6 (IL-6): Secretada por múltiples células, dentro de ellas células inmunes, fibroblastos, células endoteliales, músculo esquelético y tejido adiposo, se trata de una citoquina pleiotrópica con múltiples efectos, que van desde la inflamación y la defensa hasta el daño tisular (4). Circula de forma glicosilada, en tamaños que oscilan entre los 22 κ a 27 κ y de su receptor, homólogo al receptor de leptina, existe una forma transmembranal y otra soluble. Un complejo conformado por el receptor y por dos moléculas homodimerizadas comienza la señalización intracelular de IL-6, 1/3 de la cual se expresa en los adipocitos y en la matriz del tejido adiposo. Su expresión y secreción son de dos a tres veces mayores en el tejido visceral que en el tejido subcutáneo, circula en altos niveles sanguíneos y su expresión y niveles circulantes se correlacionan positivamente con obesidad, con intolerancia a la glucosa y con insulino resistencia. Disminuye también la señalización de insulina en tejidos periféricos, inhibe la adipogénesis y desciende la secreción de adiponectina (30). Como en el sistema nervioso central los efectos de la IL-6 son diferentes y sus niveles se correlacionan negativamente con la masa grasa en los pacientes con sobrepeso, se sugiere una deficiencia central en la obesidad (30). Adipsina y proteína estimulante de acetilación: Es una serina proteasa secretada conjuntamente con el factor D del complemento y de su interacción con los factores C3 y B del complemento se origina la proteína estimulante de la acetilación (ASP por sus siglas en inglés), proteína que afecta el metabolismo lipídico y el de la glucosa (4). Ambas proteínas se correlacionan positivamente con adiposidad, con insulino resistencia, con dislipidemia y con enfermedades cardiovasculares (30). ASP promueve la captación de ácidos grasos por incremento en la actividad de la lipoproteina lipasa, promueve la síntesis de triglicéridos por aumento de la actividad de la diacilglicerol acil transferasa y desciende la lipólisis y la liberación de ácidos grasos libres en los adipocitos (30). También incrementa el transporte de glucosa en los adipocitos, incrementando la translocación de sus transportadores y mejora la secreción de insulina estimulada por glucosa. Un receptor para ASP unido a proteína G y conocido como C5L2, se expresa en los adipocitos (30). Visfatina: Fue identificada hace varios años como un factor estimulador de colonias de células pre B y los estudios preliminares sugieren que sus concentraciones están incrementadas en humanos con obesidad abdominal, con diabetes mellitus, o con ambas. Circula en concentraciones por debajo de las de insulina (<10% en ayuno y <3% en estado prandial), considerándose muy pequeño su papel en la capacidad de mantener la normo glicemia. Como su expresión no es regulada por la alimentación ni por el ayuno, se considera un factor poco probable en la vía de señalización del receptor de insulina. Existen aspectos pendientes por aclarar, como el hecho que los niveles séricos de visfatina están variablemente correlacionados con la diabetes tipo 2 y con otros estados de insulino resistencia. El incremento de su síntesis, asociado con obesidad, podría ser una repuesta compensatoria para tratar de mantener la glicemia (31). Se ha demostrado que la visfatina tiene actividad enzimática de nicotinamida fosforibosiltransferasa, con residencia en el núcleo y el citosol, no estando claro si hay una secreción regulada de visfatina o si sus niveles séricos reflejan un escape desde las células muertas o dañadas (31). Son varios los receptores que regulan su síntesis, la cual es estimulada por glucocorticoides e inhibida por TNF-α, por IL-6, por hormona de crecimiento y por agonistas de receptor β-adrenérgico (32,33). Terapéuticamente, los agonistas PPAR gamma muestran efectos diferentes sobre su modulación y se ha demostrado recientemente que el fenofibrato estimula su producción. Omentina: Se trata de a otro péptido secretado principalmente por la grasa visceral y contrario a la visfatina, parece que se produce más en células estromales vasculares dentro de la grasa, que en los mismos adipocitos. Aunque el mecanismo de acción de la omentina, incluyendo los tejidos blancos, su receptor y sus vías de transducción, están por esclarecerse, es interesante resaltar que la omentina se produce en cantidades considerables en el tejido adiposo de humanos y de macacos, pero no en el de ratones (34). Apelina: Es un ligando del receptor huérfano AJP que actúa básicamente de forma paracrina y secretado como un prepropéptido de 77 aminoácidos que da origen a péptidos de 36, de 17 y de 13 aminoácidos, siendo los dos últimos las formas más activas (31). La única forma de metabolismo que se le conoce es a través de la enzima convertidora de angiotensina 2, limitada principalmente al endotelio de arterias, arteriolas y vénulas del corazón, riñón, epitelio tubular renal y testículos, la cual cliva la fenilalanina terminal de las adeninas 13 y 16 y las convierte en péptidos inactivos (35). El receptor de apelina se expresa ampliamente en el cerebro y en casi todos los tejidos periféricos, especialmente en células endoteliales a nivel de vasos intramiocárdicos, renales, pulmonares, adrenales y endocárdicos (31). La apelina produce vasodilatación dependiente de endotelio, mediada por óxido nítrico y vasoconstricción independiente de endotelio, por su acción sobre las células musculares lisas vasculares. Siendo la primera acción la que prima (31). Se ha descrito también el su papel en angiogénesis, el incremento en la contractilidad miocárdica, pero sin aumento del gasto cardíaco (debido a la venodilatación que disminuye la precarga) y sin producción de hipertrofia a largo plazo. Produce también inhibición en la secreción de hormona antidiurética, lo que le daría una función en el manejo de falla cardiaca (36). Sobre las hormonas pituitarias se ha visto luego de su administración: aumento de ACTH y cortisol, supresión de prolactina, FSH, LH y TSH y un efecto neutro sobre la hormona de crecimiento (31). Como adipoquina se produce de manera proporcional con la cantidad de grasa y tiene propiedades anorexígenas, acompañadas de aumento de la temperatura corporal y la actividad locomotora. Además, inhibe la secreción de glucosa dependiente de insulina (37). Resistina: Es una proteína dimérica de 108 aminoácidos con acción hiperglucemiante, conocida también como FIZZ3 (análogo 3 de la familia de moléculas FIZZ - found in inflammatory zone) y perteneciente a la familia de proteínas ricas en cisteína en el dominio C-terminal. Su expresión es quince veces mayor en el tejido adiposo visceral. Se identificó como un producto secretado por adipocitos de ratón, reprimido por las tiazolidindionas y con niveles elevados en muchos modelos múridos de obesidad; los modelos de ganancia y de pérdida de la función en roedores soportan el papel de la resistina en el incremento de la liberación hepática de glucosa (38,39). Como con la adiponectina, también hay varias formas multiméricas de resistina que circulan en el plasma y su acción celular parece depender de las formas con menor peso molecular, que son dímeros unidos por un puente disulfuro (40). Existe considerable controversia alrededor del papel de esta proteína en los humanos, debido a que la homología entre la resistina humana y la múrida no alcanza el 60%. Numerosos estudios en humanos han fallado en demostrar una correlación clara y consistente entre la expresión de resistina en el tejido adiposo, o entre los niveles circulantes de resistina y la adiposidad o la insulino resistencia (41). Pero lo que sí parece probado es que constituye un vínculo con el entorno inflamatorio, debido a su predominante producción por monocitos y por su correlación con los niveles de IL-6 (42). Proteína 4 ligante de retinol (RBP-4): Es un miembro de la superfamilia lipocalina y está regulada por cambios en los niveles del transportador 4 de glucosa (GLUT-4) en los adipocitos. Su expresión aumentada desmejora la acción de la insulina en músculo y en hígado, de forma tal que sus niveles elevados se asocian con insulino resistencia en humanos obesos y en aquellos con diabetes tipo 2, así como en pacientes delgados no diabéticos, pero con historia familiar de diabetes (43). Su mecanismo de acción no ha sido dilucidado y no está claro si el proceso compromete un ligando retinoide, o algún otro mecanismo (44). Ácidos grasos no esterificados (NEFA): Conocidos desde tiempo atrás como productos de secreción del adipocito, se liberan primariamente durante el ayuno como fuente de nutrición para el resto del cuerpo, con acciones adicionales en la homeostasis de la glucosa. Como en los adipocitos la lipólisis es reprimida por insulina, la insulino resistencia está asociada con lipólisis y con liberación de NEFA en la circulación. Un nivel elevado de NEFA inhibe la habilidad de la insulina para promover, tanto la toma periférica de glucosa en músculo y en grasa, como para reducir la producción hepática de glucosa. Los niveles elevados de NEFA de manera transitoria, como ocurre durante una comida, tienden a aumentar la secreción de insulina, mientras que las elevaciones crónicas, como en los casos de insulino resistencia, tienden a reducir la secreción de esta hormona. (45,46). El efecto neto de estas acciones es promover la utilización de lípidos como una fuente de energía para la mayoría de tejidos, mientras que para las neuronas y para los glóbulos rojos, que dependen de la glucosa como fuente de energía, se reservan los carbohidratos. Se han propuesto varios mecanismos para explicar estos efectos, dentro de ellos la activación de protein quinasa C (PKC) (47,48), el estrés oxidativo (49), la formación de ceramida, (50) y la activación del sistema inmune innato (51,52) vías estas que no se excluyen entre sí y que probablemente funcionen de manera interdependiente. Debido a que la lipólisis en los adipocitos es reprimida por la insulina, la insulino resistencia de cualquier origen puede llevar a la elevación de NEFA, la cual, a su vez, induce insulino resistencia adicional, como parte de un círculo vicioso (53). Este efecto está mediado por varios mecanismos, incluyendo la apoptosis inducida por lipotoxicidad en las células de islotes. En las células beta que escapan de la apoptosis, los NEFA descienden la "percepción" de glucosa mediante una inducción de la expresión de la proteína desacopladora 2, la cual desciende el potencial de membrana mitocondrial, la síntesis de ATP y la secreción de insulina (54). Inhibidor del activador del plasminógeno (PAI-1): Esta proteína, inhibidor primario de la formación de trombo por inhibición del activador del plasminógeno, es un miembro de la familia de inhibidores de serina proteasa y está implicada también en una variedad de procesos biológicos que incluyen la angiogénesis y la aterogénesis. Se expresa en múltiples tipos celulares dentro del tejido adiposo, su expresión y secreción son mayores en el tejido visceral que en el tejido subcutáneo y sus niveles elevados, observados en la obesidad y en la insulino resistencia, se correlacionan positivamente con el síndrome metabólico. La pérdida de peso, la restricción calórica, el ejercicio y la mejoría de la sensibilidad a la insulina, con un tratamiento a base de metformina o de tiazolidindiona, reduce significativamente sus niveles circulantes (29). Sistema renina-angiotensina-aldosterona: Varias de las proteínas de este sistema, entre ellas la renina, el angiotensinógeno, la angiotensina I, la angiotensina II, los receptores de angiotensina tipo 1 (AT1) y tipo 2 (AT2), la enzima convertidora de angiotensina y otras proteasas capaces de producir angiotensina II (quimasa, catepsina D y G)(55), son producidas en el tejido adiposo. La expresión de angiotensinógeno, de enzima convertidora de angiotensina y de receptores AT1, es mayor en el compartimiento visceral que en el subcutáneo (55) y la adiposidad se correlaciona con la angiotensina plasmática, con actividad de renina plasmática, con actividad plasmática de enzima convertidora de angiotensina (56). El angiotensinógeno es producido principalmente en el hígado y su mayor fuente extrahepática es el tejido adiposo. Un aumento en su producción contribuye a un incremento de masa adiposa, porque según se cree, la angiotensina II actúa localmente como factor trófico para la formación de nuevo tejido adiposo (57). De otra parte, la angiotensina II, que se une a sus receptores en adipocitos, en células estromovasculares y en terminales nerviosas, afectando la fisiología del tejido adiposo por alteración del flujo sanguíneo y por la actividad del sistema nervioso simpático, es capaz de promover directamente el crecimiento y la diferenciación del adipocito al promover la lipogénesis, e indirectamente, al estimular la síntesis de prostaglandinas; además, la angiotensina II regula la expresión de factores endocrinos tisulares como prostaciclina, como óxido nítrico, como PAI-1 y como leptina (56) y, la proveniente de adipocitos maduros, inhibe el reclutamiento posterior de preadipocitos. 11 β-dehidroxiesteroide dehidrogenasa: Esta enzima, de la que se han descubierto dos isoformas, cada una con propiedades y con roles biológicos únicos y localizadas ambas en el retículo endoplásmatico, cataliza la interconversión del cortisol activo a cortisona inerte. La isoforma 1, que regenera el cortisol metabolicamente activo de la cortisona, está incrementada en los sujetos obesos y la isoforma 2, que inactiva el cortisol, protege tejidos claves. Varias observaciones asocian la actividad de la enzima tipo 1 con obesidad, con insulino resistencia y con otros hallazgos de síndrome metabólico (58), sin embargo, no se han encontrado diferencias en su actividad entre pacientes obesos con diabetes tipo 2 y sus controles obesos, lo que sugiere que su desregulación se asocia más con obesidad que con diabetes de tipo 2 (59). Implicaciones fisiopatológicas Homeostasis de glucosa: A pesar de la ingesta intermitente de carbohidratos en la dieta, los niveles de glucosa sanguínea permanecen relativamente estables durante el día, lo que requiere de acciones concertadas de diferentes tejidos. Como respuesta a la elevación de glucosa que ocurre después de comer, las células pancreáticas secretan insulina, la cual promueve el depósito de glucosa en el tejido adiposo y en el músculo, e inhibe la producción de glucosa por el hígado, por supresión de la glucogenólisis y de la gluconeogénesis. De otra parte, los bajos niveles de de insulina en el estado de ayuno, combinados con una elevación de las hormonas contrarregulatorias como el glucagón, la adrenalina y los corticosteroides, promueven la producción hepática de glucosa. Recientemente ha surgido evidencia acerca del cerebro como coordinador de muchos de estos efectos, por medio de la percepción directa e indirecta de glucosa y de la información a órganos periféricos (60). Inicialmente, la idea de que el tejido adiposo pudiese tener un considerable efecto en el control glucémico global no fue fácil de aceptar y los estudios iniciales determinaron que el tejido adiposo sólo representaba una fracción de la distribución después de una comida (cerca del 10% al 15%), siendo captada por el músculo la mayor parte (61). Sin embargo, se fue haciendo evidente que las alteraciones en la adiposidad tenían profundas implicaciones en la homeostasis de la glucosa: demasiada grasa (obesidad) y muy poca (lipodistrofia) se asociaban con insulino resistencia y con hiperglucemia. Adicionalmente, los ligandos de PPARγ tenían una actividad antidiabética excelente, a pesar de que la mayor parte de ellos se encontraban en el tejido adiposo y no en el músculo. Ahora, ya se entiende que el profundo efecto de los adipocitos en el balance de glucosa está mediado por varios mecanismos, dentro de ellos el papel vital de las adipoquinas, ya sea ejerciendo efectos antihiperglucémicos como lo hacen la leptina, la adiponectina, la visfatina y la omentina, o bien, con efectos pro hiperglucémicos como lo hacen la resistina, el TNF-α, la IL-6 y el RBP4. La leptina tiene capacidad en el ratón ob/ob de revertir la hiperglucemia, incluso antes de corregir el exceso de peso y mejora la homeostasis de glucosa en el ratón lipodistrófico y en humanos con lipodistrofia, o con deficiencia congénita de leptina. Los resultados no son los mismos en el humano con obesidad típica, lo que estaría acorde con la resistencia a la leptina vista en estos pacientes (62). Las acciones anti hiperglucémicas de la leptina están mediadas por diferentes órganos. Es así como mejora la insulino sensibilidad, al disminuir los lípidos en el músculo esquelético, en el hígado y en las células beta pancreáticas, por una activación directa de la proteína quinasa activada por AMP y por la inhibición de la malonil coenzima-A y por acciones indirectas mediadas en las vías neurales simpáticas, estimulando receptores adrenérgicos que incrementan el transporte de los ácidos grasos a la mitocondria para la beta oxidación (63). También existe discusión acerca de la existencia de un eje adiposo-insulina, en el cual la insulina promueve la secreción de leptina y la leptina a su vez, inhibe la liberación de insulina, debido a la inhibición de la síntesis de proinsulina y a una reducción de su secreción. Consistente con esta idea está el hecho de que con la ablación de los receptores de leptina de las células beta, se da un incremento en la secreción basal de insulina (64). La adiponectina mejora la sensibilidad a la insulina, desciende el flujo de ácidos grasos libres e incrementa su oxidación, inhibe las principales enzimas gluconeogénicas hepáticas, reduce la liberación hepática de glucosa y en el músculo, estimula el uso de glucosa y la oxidación de los ácidos grasos, efectos que en parte están mediados por la activación de AMPK(4). Además, favorece el desplazamiento de los transportadores de glucosa, sobre todo de Glut -4, a la superficie. Los modelos de pérdida de función de adiponectina han mostrado resultados variables, sin que se conozca el porqué (14). La adiponectina no tiene efecto en la secreción de insulina en los islotes de ratones sanos o en humanos, pero si aumenta la secreción de insulina mediada por glucosa, en los ratones con obesidad inducida por dieta. Algunos estudios sugieren que individuos con altos niveles de adiponectina tienen un menor riesgo para desarrollar diabetes que aquellos con bajos niveles (65). La adiponectina plasmática desciende antes del comienzo de la obesidad y de la insulino resistencia, lo que sugiere que un estado de hipoadiponectinemia contribuye a la patogénesis de esas condiciones (66). Sus niveles se incrementan, así como la expresión de los genes para su síntesis, cuando mejora la sensibilidad a la insulina luego de disminución de peso, de restricción calórica y de tratamiento con drogas sensibilizadoras de insulina (67), dentro de estas las tiazolidindionas, que actúan uniéndose al elemento de respuesta de PPARγ en la región promotora de adiponectina (68). La visfatina, que se une y activa el receptor de insulina en un sitio diferente al de la insulina, estimula la fosforilación del mismo y la de los substratos 1 y 2 del receptor (IRS-1, IRS-2), la unión de la fosfoquinasa 3 (PI3K) a IRS-1 e IRS-2 y la fosforilación de señales de las quinasas Akt y MAPK. En adición, la visfatina estimula la diferenciación de preadipocitos a células maduras, promueve la acumulación de triglicéridos, acelera la síntesis de triglicéridos desde glucosa e induce la expresión de genes que codifican para PPARγ, para sintasa de ácidos grasos, para diacilglicerol aciltransferasa y para adiponectina y mediante una reducción en la liberación de glucosa de los adipocitos y el estímulo de su utilización en la periferia, disminuye también la glucemia (31). La última de las adipoquinas con efecto positivo sobre la homeostasis de glucosa es la omentina, que al igual que la visfatina tiene efectos positivos en la captación de glucosa, aunque funciona como un sensibilizador de insulina y tiene propiedades insulino miméticas (69). Los niveles de TNF-α están elevados en obesidad y en otros estados de insulino resistencia como la sepsis; la adición de TNF-α a las células reduce la acción de la insulina y su bloqueo, por medios bioquímicos o genéticos, restablece la insulino sensibilidad tanto in vivo como in vitro. Este efecto es mediado por la activación de serina quinasas que incrementan la fosforilación de serina en IRS1 e IRS2, convirtiéndolos en pobres sustratos para las quinasas del receptor de insulina y que aumentan su degradación. También altera la señalización de insulina indirectamente, mediante el aumento de los NEFA. En el hígado, suprime la expresión de genes comprometidos en la captación y metabolismo de glucosa y en la oxidación de ácidos grasos e incrementa la expresión de genes comprometidos en la síntesis de novo de colesterol y de ácidos grasos (4) La IL-6 disminuye la acción de la insulina en tejidos periféricos por reducción de la expresión de los componentes de señalización del receptor de insulina y por inducción del supresor 3 de señalización de citoquinas SOCS3 (regulador negativo de la señalización de leptina e insulina). Estas citoquinas pueden promover la insulino resistencia por varios mecanismos, dentro de ellos la fosforilación de IRS-1 por la c-Jun N-terminal quinasa 1 (70), la activación del NF κβ (71), la inducción del SOCS3 (72) y la producción de ROS (73). La IL-6 también influye en la tolerancia a la glucosa mediante una regulación negativa de vifastina (74). Mientras que la resistina podría estar implicada en la reducción de la captación de glucosa por los músculos y la grasa, pero con un efecto menos vigoroso que en el hígado, en donde incrementa su liberación (75), la angiotensina II inhibe la lipólisis, promueve la lipogénesis, desciende la captación de glucosa dependiente de insulina e incrementa la gluconeogénesis hepática y la glicogenólisis (56). La proteína quimio atrayente de macrófagos (MCP-1) empeora la insulina resistencia cuando se sobre expresa (76) y su ablación dirigida, o la de su receptor, reduce la infiltración por macrófagos de los depósitos grasos y mejora la sensibilidad de la insulina a pesar de no cambiar el peso corporal (77). Por su parte, los macrófagos activados secretan factores inflamatorios que contribuyen a la insulino resistencia (78). La última de las adipoquinas que influye negativamente el metabolismo de glucosa es la RBP-4, cuya sobre expresión desmejora la acción de la insulina en el músculo y en el hígado. Sus niveles elevados se asocian con insulino resistencia en humanos obesos, en aquellos con diabetes tipo 2, así como en pacientes delgados no diabéticos, pero con historia familiar de diabetes. Su mecanismo de acción no está dilucidado y no es claro si el proceso compromete un ligando retinoide, o algún otro mecanismo (79). Balance de energía: Durante mucho tiempo se ha considerado que el balance de energía en los animales refleja la relación entre el consumo de energía, el gasto energético y la energía almacenada, expresados como ecuación en la primera ley de la termodinámica, siendo, entonces, el tejido adiposo la representación de un exceso de consumo de energía en relación con el gasto. Aunque en principio es cierto, esta representación subestima los hallazgos claves de la homeostasis in vivo, porque aunque el consumo de alimentos es relativamente fácil de medir, no es el parámetro preciso que determina la cantidad de energía que entra en el sistema, ya que en general, se ignora la eficiencia de absorción de calorías en el intestino. Otra consideración es que la respuesta del cuerpo a las alteraciones en el ingreso o en el gasto de energía no es estático y en general, la homeostasis de energía se regula para defender el más alto peso alcanzado, de forma tal, que reducciones voluntarias en el consumo de alimentos, son contra reguladas por reducciones involuntarias en el gasto de energía, haciendo que la pérdida de peso sea más difícil que la simple interpretación de la ecuación (80). Aunque el consumo calórico está representado, casi en su totalidad, por el consumo de alimentos (menos lo que falla en ser absorbido), el gasto de energía tiene más componentes, incluyendo el metabolismo basal, la actividad física (voluntaria e involuntaria) y la termogénesis adaptativa. Esta última categoría incluye la pequeña cantidad de energía empleada en la absorción y en el procesamiento de la dieta (termogénesis inducida por dieta), así como la energía empleada para mantener la temperatura corporal durante el frío. Una de las principales adipoquinas comprometida en la regulación de este balance energético y el mejor adipostato, al reprimir el consumo de comida y promover el gasto energético, es la leptina, secretada casi exclusivamente por la grasa. La activación de los receptores de leptina en cuanto a los núcleos arcuato, ventromedial y dorso medial del hipotálamo lleva a la represión de vías orexigénicas (comprometiendo el neuropéptido Y (NPY) y péptido agouti relacionado) y a una inducción de vías anorexigénicas (comprometiendo pro-opiomelanocortina, cocaína y transcripto regulado por anfetamina). La vía de señalización leptina-proopiomelanocortina es, además, la que aglutina el mayor número de mutaciones monogénicas, causantes de obesidad en el ser humano (81). Los efectos dependientes de leptina en el consumo alimentario y en el gasto de energía en último término, divergen parcialmente en lo referente a las señales del receptor de melanocortina 4 (MC4) en el sistema nervioso central. Recientemente se ha puesto atención a la acción de la leptina en sitios no hipotalámicos, como el tallo cerebral caudal, el músculo y las células beta pancreáticas (82). Periféricamente, esta adipoquina modula la acción de la quinasa dependiente de AMP (AMPK), sensor energético de la célula, encargado de la puesta en marcha o de la inhibición, de los productos anabólicos y catabólicos celulares. El efecto de la leptina con relación a la homeostasis energética, como señal de suficiencia, más que como señal antiobesidad, está bien documentado, así como los cambios adaptativos que genera en respuesta a la ingesta de calorías: inhibición del apetito (mediante inhibición del NPY y estimulación en la producción de propoiomelanocortina en el núcleo arcuato hipotalámico) e incremento del gasto energético (por medio de la estimulación del sistema nervioso simpático). Sin embargo, en situaciones de exceso de reservas energéticas y por lo tanto, de leptina, como es el caso de la obesidad, este mecanismo no se muestra tan eficiente, postulándose diversas alteraciones (tanto anatómicas como funcionales) de la vía de señalización de la leptina, situación denominada "resistencia a la leptina" (83). El descenso característico en la termogénesis durante el ayuno y durante la hiperfagia post ayuno, esta mediado, al menos en parte, por un descenso en leptina (4), de forma tal forma que la deficiencia de leptina se percibe como un estado de ayuno no mitigado, llevando a respuestas compensatorias como hiperfagia, disminución de la rata metabólica y cambios en los niveles hormonales diseñados para restablecer el balance de energía (84). La cantidad de adiposidad está regulada por vías neurales y el tejido adiposo se comunica con el sistema nervioso central por medio de una rica red de nervios periféricos que transmiten señales aferentes acerca del estado energía al cerebro, lo que se traduce en una sensibilidad incrementada a la leptina, aumentando su efecto. Los cúmulos de grasa están ricamente inervados por fibras simpáticas y la activación de ellas se asocia con un incremento de la lipólisis; esos nervios también regulan la celularidad del cúmulo graso (la denervación de depósitos específicos resulta en un incremento al doble del número de adipositos en hamster y ratas) (85). Estudios recientes sugieren que las señales neurales aferentes desde el tejido adiposo hasta el cerebro también pueden regular la adiposidad, sin que esté claro cual es el desencadenante en el cerebro, pudiendo estar incluidos reactivos intermediarios del oxígeno, niveles de ATP o aún, la producción local de calor (86). Otra vía por la que el tejido adiposo modula el balance de energía corporal es la de las alteraciones en su propio metabolismo, hecho que se comprende mejor en la grasa parda, ya que los adipocitos pardos están altamente especializados en respiración desacoplada, la cual disipa la energía química en forma de calor (87). En la dinámica mitocondrial clásica, la oxidación de energía está ligada al transporte de electrones por medio de tres complejos de proteínas que conducen al desarrollo de un gradiente electroquímico que se origina por la extrusión de protones a través de la membrana mitocondrial interna. Normalmente se disipa en el complejo V, que liga el flujo de protones con la síntesis de ATP, pero como el adipocito pardo lo que expresa es UCP-1, esta proteína le permite al flujo de protones volver a través de la membrana mitocondrial interna sin el desarrollo de una síntesis concomitante de ATP, hecho que se traduce en la generación de calor. En roedores, el tejido adiposo pardo hace una contribución sustancial al metabolismo total de energía y en ratones sin tejido adiposo, con un reducido gasto de energía, se ha observado susceptibilidad a la obesidad inducida por dieta; es interesante resaltar que los ratones carentes de UCP-1, aunque son sensibles al frío, no son obesos. Estos modelos genéticos en roedores sugieren que si bien el tejido adiposo pardo puede influenciar el metabolismo energético corporal por medio de la expresión de UCP-1, las vías no están del todo entendidas, a pesar de que sea evidente que una sobre expresión de UCP-1 en el tejido adiposo blanco, se traduce en una reducción de la adiposidad, efecto que al parecer, está mediado neuralmente (3). Otro mecanismo que también se ha propuesto como reductor de la adiposidad es el aumento en la oxidación de los ácidos grasos en el tejido adiposo blanco (sin desacoplamiento) y aunque el incremento en la lipólisis sea insuficiente para promover perdida de peso, los ácidos grasos liberados deben ser oxidados, así la oxidación sola tampoco sea suficiente, a menos que se consuma el ATP generado (88). Obesidad visceral, inflamación e insulina resistencia: El tejido adiposo de los pacientes obesos se caracteriza por hipertrofia e hiperplasia de los adipocitos y por cambios en sus funciones metabólicas, estando demostrado que el adipocito es el mayor productor de adipoquinas inflamatorias en estas condiciones (89-93). Un mecanismo mediante el cual se induce esta producción es el estrés del retículo endoplásmico (entendido como un aumento de sus demandas de funcionamiento) inducido por la obesidad, lo que ocasiona cambios en la arquitectura, aumento en la síntesis de proteínas y de lípidos y perturbaciones en los flujos de energía y de nutrientes intracelulares en el tejido adiposo (94), que llevan a la activación de diferentes vías de señalización, entre ellas, las quinasas JNK , IKK y proteína quinasa C, induciendo (por regulación post transcripcional) la producción adicional de mediadores inflamatorios, especialmente de TNF-α y de IL-6, e inhibiendo directamente la acción de la insulina a través de la transfosforilación de serina del substrato 1 del receptor de insulina (IRS-1), al igual que lo hace el TNF-α de manera directa, o a través de la inducción de transcripción y de síntesis de SOCS, que induce además, la proteólisis de dichos substratos (Figura 1) (95,96).
Recibido: Junio 4 de 2007. Aceptado: Julio 19 de 2007.
Resumen

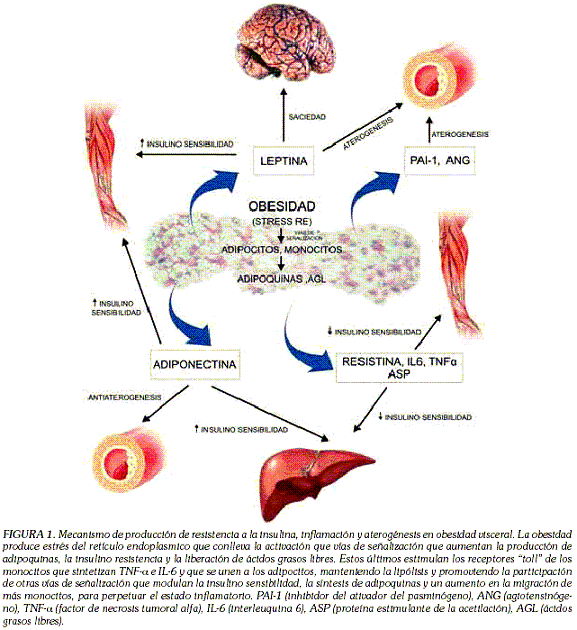
Otro mecanismo que también puede ser relevante en la iniciación de inflamación es el estrés oxidativo que se produce como consecuencia del incremento en el aporte de glucosa al tejido adiposo: al aumentar las células endoteliales la captación de glucosa, se genera un aumento en la producción de radicales super óxido a nivel mitocondrial que ocasiona daño oxidativo, incremento en la producción de citoquinas inflamatorias (97) y activación de las cascadas de señalización inflamatoria dentro de la célula endotelial (Figura 1) (98).
Como los adipocitos de pacientes obesos también presentan una menor densidad de receptores de insulina y una mayor de receptores beta-3 adrenérgicos, se incrementa entonces la tasa de lipólisis con liberación de ácidos grasos libres, situación que tiene varias consecuencias metabólicas, dentro de ellas el aumento en la producción de radicales libre de oxígeno, la inducción de insulino resistencia, el sinergismo en la acción de la IL-6 y el TNF-α y la inducción de apoptosis en la célula beta pancreática, efectos todos categorizados como de lipotoxicidad (17). Como consecuencia de los defectos inducidos en la señalización de la insulina hay potenciación de la lipólisis y disminución de la síntesis de adiponectina.
Además de las mencionadas, la infiltración por macrófagos es otra de las características del tejido adiposo en obesidad: a través de una comunicación entre los macrófagos y los adipocitos se establece el estado inflamatorio que caracteriza a la obesidad y que se inicia con la unión de los ácidos grasos libres a los receptores Toll de los monocitos, lo que lleva a la activación de vías de señalización como las quinasas reguladas por señales extracelulares (ERK y JK), que a su vez inducen la producción de TNF-α e IL-6, que al unirse a sus receptores en adipocitos inducen de nuevo vías de señalización en el adipocito (como ERK, PK y p38k), terminando en la síntesis de la proteina-1 quimioatrayente de monocitos (AP-1), en un aumento en la migración de los mismos y en la perpetuación del estado inflamatorio. Los niveles aumentados de TNF-α, como consecuencia de la comunicación entre adipocito y monocito, induce la producción de otros factores inflamatorios que aumentan la lipólisis e inhiben la síntesis de adiponectina (17). Hay que recordar que los adipocitos y los macrófagos comparten varias características, dentro de ellas los productos de expresión de sus genes y su capacidad funcional, ya que los macrófagos pueden almacenar lípidos y los pre adipocitos exhibir propiedades fagocíticas y antimicrobianas, llegando a poder diferenciarse en macrófagos (99,100).
Patología cardiovascular y aterogénesis: El proceso de aterosclerosis se reconoce como un proceso inflamatorio en el que los monocitos, se adhieren al endotelio y migran al espacio subendotelial para convertirse en células espumosas, con lipoproteínas asociadas. La producción de metaloproteinasas por las células espumosas lleva a la ruptura de la capa fibrosa de la placa y a la ruptura de la placa en sí (101). En este proceso inflamatorio las adipoquinas juegan diversos papeles: el TNF-α activa el NF-κβ con los subsecuentes cambios inflamatorios en el tejido vascular, entre ellos el aumento en la expresión de la molécula de adhesión intracelular y de la molécula de adhesión de células vasculares que incrementan la adhesión de los monocitos a la pared del vaso, con una mayor producción de MCP-1 y MCSF en las células endoteliales y en las células de músculo liso vascular y con un aumento también en la expresión de óxido nítrico sintetasa inducible (ιNOS), de citoquinas y de súper oxido dismutasa (4).
Por otra parte, la estimulación simpática que desencadena la leptina favorece el incremento de la tensión arterial, mientras que el ambiente inflamatorio que generan las quimioquinas y citoquinas de la pared vascular y la síntesis de factores procoagulantes como el inhibidor del plasminógeno tisular (PAI-1), principalmente por la grasa visceral, incrementan el riesgo de patología coronaria. En este aspecto hay que destacar el potencial papel patológico del TNF-α que se produce en el tejido adiposo periarteriolar, que, en un efecto paracrino, determina un descenso en la producción de óxido nítrico que ocasiona una vasoconstricción arteriolar mantenida. Estudios in vitro e in vivo demuestran que la leptina tiene actividad angiogénica, que contribuye a la trombosis arterial a través del receptor plaquetario de leptina y que in vitro, estimula la producción de especies reactivas de oxígeno como resultado de la activación de monocitos (102-104). La leptina, especialmente en presencia de altos niveles de glucosa, estimula también a los macrófagos a acumular colesterol (105). La IL-6, que ejerce actividad pro inflamatoria mediante el incremento de IL-1 y de TNF-α, estimula también la producción hepática de proteína C reactiva, considerada como factor predictor de arteroesclerosis (105). PAI-1, que inhibe la ruptura de coágulo de fibrina favoreciendo la formación de trombos sobre las placas arteroscleróticas rotas, presenta concentraciones anormalmente altas en hiperglucemia, en hipertrigliceridemia y en obesidad (106,107). Como en humanos los niveles de PAI-1 se correlacionan con eventos arteroscleróticos y con mortalidad, se lo ha sugerido como un factor de riesgo independiente para enfermedad coronaria (108).
El angiotensinógeno también juega su papel en la patología cardiovascular, pues al estimular la molécula de adhesión intracelular (ICAM), la molécula de superficie de adhesión celular (VCAM-1), la proteína quimiotáctica para monocitos (MCP-1) y el factor estimulador de colonias de monocitos (M-CSF) en las paredes vasculares, reduce la biodisponibilidad de óxido nítrico, con una pérdida en la capacidad vasodilatadora y con un incremento de la adhesión plaquetaria a la pared vascular (4).
En pacientes asintomáticos con una historia familiar de enfermedad coronaria, los niveles plasmáticos de resistina son predictores de arterosclerosis. La resistina activa las células endoteliales in vitro y cuando se incuban con resistina humana recombinante, liberan más endotelina 1 y más VCAM 1. También se ha reportado que es capaz de inducir proliferación de las células de músculo liso a nivel aórtico, aumentar la expresión en las células endoteliales de mRNA de VCAM, de ICAM-1 y depentraxina-3, expresando entonces un patrón bioquímico de disfunción endotelial (4, 109,110).
Entre los hallazgos que soportan el papel endocrino de la MCP-1 en el desarrollo de arteroesclerosis se ha visto, en el modelo múrido, incrementos en el número de monocitos circulantes, en la acumulación de monocitos en las arterias colaterales y en la formación de íntima (111).
La angiotensina II por su parte, media el incremento del tono vascular, la secreción de aldosterona y la reabsorción de agua, todos actuando en la regulación de la presión sanguínea, de forma tal que el sistema renina angiotensina aldosterona puede ser el puente entre la hipertensión y la obesidad (30). En oposición, la adiponectina ejerce múltiples efectos benéficos (antiaterogénicos), regulando el tono vascular, mejorando el perfil lipídico y contrarrestando la arterogénesis y los riesgos de ruptura y de desprendimiento de la placa de ateroma por diversos mecanismos, entre ellos el de de la regulación negativa de los receptores scavenger (30) y el de activación de las células endoteliales por medio de una reducción en la producción de moléculas de adhesión y de la inhibición del factor de TNF-α y NF κβ (112). Dentro de la pared vascular la adiponectina también inhibe la adhesión monocitaria (al descender la expresión de moléculas de adhesión), la transformación de macrófagos en células espumosas (al inhibir la expresión de receptores scavenger) y disminuye la proliferación de las células migrantes de células de músculo liso, en respuesta a factores de crecimiento; adicionalmente, incrementa la producción de óxido nítrico (NO) en las células endoteliales. Estos efectos son mediados por un incremento de la fosforilación del receptor de insulina, por activación de AMPK y por modulación de la vía del NF κβ.
Aunque se requiere de más estudios para clarificar si la adiponectina predice o no, de forma independiente, el riesgo de eventos coronarios, en hombres con diabetes tipo 2 los niveles incrementados de adiponectina se relacionan con disminución del riesgo de enfermedad coronaria, en una asociación que parece estar mediada por los efectos de la adiponectina en el HDL, al incrementarlo (113) y en contraposición con estudios en indígenas americanos, en los que la adiponectina no se relaciona con la incidencia de enfermedad coronaria (114).
Neoplasias: Se ha postulado la posible implicación de la leptina y de la adiponectina en la aparición y progresión de determinados tipos de neoplasias, con efecto estimulador e inhibidor de la mitosis, respectivamente. En pacientes con altos grados de inflamación debida cáncer, lL-6 se relaciona con una mayor insulino resistencia (115).
Otros sistemas hormonales: En adición a sus efectos en la homeostasis de energía, la leptina regula la función neuroendocrina y sistemas endocrinos tradicionales. La deficiencia de leptina se asocia con activación del eje hipotálamo-hipófisis-adrenal en el modelo múrido (su papel sobre el eje humano está esclarecerse) (116) y con supresión del eje hipotálamo-hipófisis-tiroides, puesto que normalmente estimula la expresión de TRH y la secreción de TRH desde las neuronas hipotalámicas y del eje hipotálamo-hipófisis-gónada. Su administración durante el ayuno previene los cambios inducidos en estos ejes en el hombre sano (117) y por medio de los receptores periféricos de leptina en ovarios, en testículos, en próstata y en placenta, también tiene efectos directos (118).
Otros efectos: Otros importantes efectos endocrinos de la leptina incluyen la hematopoyesis, al promover la proliferación y diferenciación de las células hematopoyéticas; la regulación de la función inmune, normalizando la función inmunológica suprimida asociada a malnutrición y a deficiencia de leptina, e influenciando producción de citoquinas por las células inmunes; la angiogénesis, estimulando el crecimiento de las células endoteliales y acelerando la cicatrización de heridas y por último el desarrollo óseo, por sus efectos antiosteogénicos que al parecer están mediados por neuronas del núcleo ventral medial del hipotálamo, que influencian la actividad del sistema nervioso simpático (119, 120).
Implicaciones terapéuticas
La gran cantidad de vías por medio de las cuales el tejido adiposo controla el peso corporal, la glucemia y otras funciones metabólicas, ha llevado a la idea de que la manipulación de la biología del adipocito puede ser una estrategia terapéutica útil en el control de ciertas enfermedades metabólicas. A pesar de esta clara conexión, está claro también que la disminución en el número de células grasas no es una estrategia viable para promover salud y que la remoción del tejido adiposo no mejora, ni en animales obesos ni en humanos, los parámetros metabólicos. Es más, los sujetos con lipodistrofia presentan insulino resistencia, hiperlipidemia y esteatohepatitis, que a menudo lleva a cirrosis, asociada con un depósito ectópico de lípidos. Una estrategia mucho más fisiológica sería la de manipular la biología del adipocito en vías que promovieran la salud. A este respecto, algunas opciones terapéuticas serían: administración de leptina y adiponectina recombinante, análogos de visfatina, bloqueadores de PAI-1 y de 11 β-HSD y aumento de la excreción de RBP4 (121). Se están desarrollando también anticuerpos bloqueantes de TNF-α o de IL-6, inhibidores de PAI-1 y reguladores del sistema renina-angiotensina-aldosterona. La inhibición de este sistema, bien con inhibidores de enzima convertidota, o bien con bloqueadores de receptor AT1, desciende el peso y mejora la sensibilidad a la insulina en ratones (122). Aunque algunos estudios aleatorios han demostrado la capacidad de los ACE para disminuir la incidencia de diabetes mellitus tipo 2, el efecto de la inhibición del sistema en la sensibilidad a la insulina ha dado resultados contradictorios (123,124).
La contribución del tejido adiposo pardo al balance total de energía en los humanos está inexplorada y se sabe que el tejido pardo que se observa alrededor de los grandes vasos del área interescapular de los niños, desaparece con la madurez. Los individuos con niveles crónicamente elevados de catecolaminas, como son los pacientes con feocromocitomas, desarrollan depósitos de tejido adiposo pardo, así como lo hacen los sujetos crónicamente expuestos a bajas temperaturas. La reactivación del tejido pardo (o promoción de un fenotipo similar al pardo en el adipocito blanco) puede ser una estrategia terapéutica viable en humanos (3). También es probable que la genómica nutricional lleve a la posibilidad de individualizar los consejos dietéticos basándose en el perfil genético (polimorfismos específicos), de tal forma que se pueda modular la producción de adipoquinas para disminuir el riesgo.
Las líneas de investigación en desarrollo se dirigen hacia dos áreas fundamentales. Por una parte, al incremento de la termogénesis, por medio de la influencia sobre el tejido adiposo pardo, sus receptores para catecolaminas y las proteínas desacopladoras (UCP) implicadas en la disipación de energía en forma de calor y por otro lado, a la inhibición de la lipogénesis, con especial atención en la señalización de la insulina y de los receptores endocanabinoides.
Dentro del arsenal terapéutico con que se cuenta actualmente, hay dos medicamentos, las tiazolidindionas y la metformina, que han demostrado algunas acciones sobre el metabolismo del tejido adiposo. Las primeras, produciendo disminución de leptina, de resistina, de TNF-α, de PAI-1, de RBP4, de MCP-1, de la infiltración de macrófagos en el tejido adiposo (125) y de la actividad del sistema renina angiotensina y la segunda, produciendo disminución de leptina, de IL-6, de PAI 1 y aumentando la adiponectina (aunque no en todos los estudios) y la resistina. En conclusión y a pesar de la falta de opciones terapéuticas basadas en la fisiología del adipocito que hay en el momento, el conocimiento cada vez mayor de su fisiología permitirá en el futuro una mejor manipulación del tejido adiposo y una disminución de las patologías altamente mórbidas que le acompañan.
Referencias
1. Stiteri PK1. Adipose tissue as a source of hormones. Am J Clin Nutr . 1997; 5:277-282 [ Links ]
2. Zhang Y, Proenca R, Maffel M, Barione M, Leopold L, Friedmand JM. Positional cloning of the Mouse obese gene and its human homologue. Nature. 1994; 372:425 - 432. [ Links ]
3. Rosen E.D. Spiegelman B.M. Adipocytes as regulators of energy balance and glucosa homeostasis. Nat 2006; 444(5483): 848-853. [ Links ]
4. Ronti T., Lupattelli G., Mannarino E. The endocrine function of adipose tissue: an update. Cliln Endocrinol 2006; 64:355-365. [ Links ]
5. Yki-Jarvinen,H. Ectopic fat accumulation: an important cause of insulin resistance in humans. Journal of the Roya Society of Medicine. 2002; 95:39-45. [ Links ]
6. Trayhurn,P. Endrocrine and signalling role of adipose tissue: new perspectives of fat. Acta Physiol Scan. 2005; 184: 285-293. [ Links ]
7. Friedman, J. M. Leptin and the regulation of body weight. Harvey Lect. 1999; 95, 107-136. [ Links ]
8. Friedman, J. M. The function of leptin in nutrition, weight, and physiology. Nutr. Rev. 2002; 60: S1-S14. [ Links ]
9. Halaas J. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995; 269: 543-546 [ Links ]
10. Pelleymounter M Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995; 269: 540-543 [ Links ]
11. Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokine by adipose tissue, adipose tissue matrix and adipocytes form visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology. 2004;145:2273-2282. [ Links ]
12. Bjorbaek C, Kahn BB Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res. 2004; 59:305-331. [ Links ]
13. Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J. Identification and expression cloning of a leptin receptor Cell 1995;83: 1263-1271. [ Links ]
14. Argente J, Martos-Moreno G.A., Hernandez M. Mesa redonda: El tejido adiposo como glándula endocrina. Obesidad y síndrome metabólico. Bol Pedriatr 2006; 46: 269-274. [ Links ]
15. Schwartz, M. W., Seeley, R. J., Campfield, L. A., Burn, P. & Baskin, D. G. Identification of targets of leptin action in rat hypothalamus. J. Clin. Invest 1996; 98: 1101-1106. [ Links ]
16. Bjorbaek, C. & Kahn, B. B. Leptin signaling in the central nervous system and the periphery. Recent Prog. Horm. Res 2004; 59: 305-331. [ Links ]
17. Morales, E. Síndrome X vs síndrome metabólico: Entendiendo sus coincidencias y sus directas hacia una nueva cardiología.Arch Cardiol Mex 2006; 76: S4, 173-188. [ Links ]
18. Sierra-Hongmann et al. Biological action of leptin as an angiogenic factor. Science 1998. 281: 1686-1683. [ Links ]
19. Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Walsh K. Obesity , adiponectin and vascular inflammatory disease. Current Opinion in Lipidology. 2003; 14: 561-566. [ Links ]
20. Chandran M, Phillps SA, Ciaraldi T, Henry RR . Adiponectin: more than just another fat cell hormone? Diabetes Care2003; 26:2442-2450. [ Links ]
21. Arita Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun 1999; 257: 79-83. [ Links ]
22. Yatagai T. Hypoadiponectinemia is associated with visceral fat accumulation and insulin resistance in Japanese men with type 2 diabetes mellitus. Metabolism 2003; 52: 1274-1278. [ Links ]
23. Hotta K. Circulating concentrations of the adipocyte protein adiponectin are decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes 2001; 50: 1126-1133. [ Links ]
24. Yamauchi T. Adiponectin stimulates glucose utilizacion and fatty acido oxidation by activating AMP - activated protein kinase. Nature Medicine 2002. 8: 1288-1295. [ Links ]
25. Chandran M, Phillips S.A., Ciraldi T, Henry RR. Adiponectin more than just another fat cell hormones. Diabetes Care 2003. 26: 2442-245. [ Links ]
26. Hotamisligil, G. S. The role of TNF and TNF receptors in obesity and insulin resistance. J. Intern. Med 1999; 245: 621-625. [ Links ]
27. Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004; 145:2273-2282. [ Links ]
28. Pittas AG, JOseph NA, Greenberg AS. Adipocitokines and insulin resistance. J Clin Endocrinol Metab 2004; 89: 447 -52. [ Links ]
29. Ruan H, Miles PD, Ladd CM, Toses K, Golub TR, Olefsky JM, Lodish HF 2002. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor alfa: implications for insulin resistance. Diabetes 2002; 51:3176-3188. [ Links ]
30. Kershaw E. Flier J. Adipose Tissue as an Endocrine Organ. J Clin Endocrinol Met 2004; 89(6): 2548 - 2556 [ Links ]
31. Bletowski J. Apelin and visfatin: Unique beneficial adipokines upregulated in obesity? Med Sci Monit 2006; 12 (6): 112-119. [ Links ]
32. Kralisch S, Klein J., Lossner U. Et al Hormonal regulation of the novel adipocytokine visfatinin 3T3-L1 adipocytes. J. Endocrinol 2005; 185: R1-8 [ Links ]
33. Kralisch S, Klein J., Lossner U. Interleukine-6 is a negative regulator of visfatin gene expression in 3t3L-1 adipocites. Am J Physiol endrocrinol Metabl 2005; 289:E586-90. [ Links ]
34. Yang R.Z. et al Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am.J Physiol. Endocrinol. Metab. 2006; 290: E1253-E1261. [ Links ]
35. Lee D.K., Valdivia V.R., Nguyen T et al. Modification of the terminal residue of apelin 13 antagonizes its hypotensive action. Endocrinology 2005; 146:231-36. [ Links ]
36. Lee C.R., Watkins M.L., Patterson J.H., Vasopressin: a new target for the treatment of heart failure. Am Heart J 2003; 146: 9-18. [ Links ]
37. Jaszberenyi M, Bujdoso E., Telegdy G. Behavioral, neuroendocrine and thermoregulatory actions of apelin-13. Neuroscience 2004; 129: 811-16 [ Links ]
38. Banerjee, R. R. et al. Regulation of fasted blood glucose by resistin. Science2004; 303: 1195-1198. [ Links ]
39. Banerjee, R. R. et al. Regulation of fasted blood glucose by resistin. Science2004; 303: 1195-1198. [ Links ]
40. Patel, S. D., Rajala, M. W., Rossetti, L., Scherer, P. E. & Shapiro, L. Disulfide-dependent multimeric assembly of resistin family hormones. Science 2004 304, 1154-1158. [ Links ]
41. Banerjee RR, Lazar MA . Resistin : molecular history and prognosis J Mol Med 2003; 81:218-226. [ Links ]
42. Reilly M.P., Lehrke M., Wolfe M.L., Rohatgi A., Lazar M.A., Arder D.I., Resistin is an inflammatory marker of atherosclerosis in humans1. Circulation 2005; 932-939. [ Links ]
43. Yang Q. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature2005; 436: 356-362. [ Links ]
44. Graham T.. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med 2006; 354: 2552-2563. [ Links ]
45. Roden M. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest 1996; 97: 2859-2865. [ Links ]
46. Roden M. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes 2000;49: 701-707. [ Links ]
47. Griffin M. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes1999; 48, 1270-1274. [ Links ]
48. Schmitz-Peiffer C. Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes 1997; 46, 169-178 . [ Links ]
49. Paolisso G.. Does free fatty acid infusion impair insulin action also through an increase in oxidative stress? J. Clin. Endocrinol. Metab1999; 81, 4244-4248. [ Links ]
50. Hajduch E. Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetología 2001; 44, 173-183. [ Links ]
51. Shi H. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest 2006; 116, 3015-3025. [ Links ]
52. Song M, Kim K. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem. Biophys. Res. Commun 2006; 346: 739-745. [ Links ]
53. Eldor R, Raz I. Lipotoxicity versus adipotoxicity - the deleterious effects of adipose tissue on beta cells in the pathogenesis of type 2 diabetes. Diabetes Res. Clin. Pract 2006; 74, S3-S8. [ Links ]
54. Lowell B. Mitochondrial dysfunction and type 2 diabetes. Science 2005; 307: 384-387 [ Links ]
55. Goznes G.H., Blaak E.E., van Baal M.A. Possible involvement of the adipose tissue renin-angiotensin system in the pathophysiology of obesity and obesity-related disorders. Obes Rev 2003; 4:43-55. [ Links ]
56. Engeli S, Schling P, Gorzelniak K, Boschmannn M, Janke J.. The adipose tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? Int J Biochem Cell 2003; 35: 807-825. [ Links ]
57. Ailhaud G, Fukamizu A., Massiera F., Negrel R., Saint-Marc P., Teboul M. Angiotensinogen, angiotensin II and adipose tissue development. International Journal of Obesity and Related Metabolism Disorders. 2000; 24: S33-S35. [ Links ]
58. Wake D, Rask E, Livingstone D, Soderberg S. Local and systemic impact of transcriptional up-regulation of 11ß hidroxiesteroid dehidrogenase type 1 in adipose tissue in human obesity. Journal Clin Endocrinol and Metabolism 2003; 88: 3983 -3988. [ Links ]
59. Lobby P. Changing concepts of atherogenesis. Journal of Internacional Medicina 2000; 247: 349-358. [ Links ]
60. Herman M. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J. Clin. Invest. 2006 116, 1767-1775 . [ Links ]
61. Kahn B. Glucose transport: pivotal step in insulin action. Diabetes 1996; 45: 1644-1654. [ Links ]
62. Heymsfield S. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. J. Am Med. Assoc. 1999; 282: 1568-1575. [ Links ]
63. Minokishi Y, Kim Y.B,. Leptin stimulates fatty-acid oxidation by activating AMP-activate protein kinase. Nature 2002; 415:339-343. [ Links ]
64. Kieffer T. J. The adipoinsular axis: effects of leptin on pancreatic -cells. Am. J. Physiol. Endocrinol. Metab2000; 278: E1-E14 [ Links ]
65. Spranger J, Kroke A, Mohkig M, Bergmann M.M., Ristow M. Adiponectin and protection against type 2 diabetes mellitus. Lancet 2003;361: 226 - 228 [ Links ]
66. Hota K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen BC, Matsuzawa Y. Circulating concentrations of the adipocyte protein adiponectin and decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes. 2001;50: 1126-1133 [ Links ]
67. Diez JJ, Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur J Endocrinol.2003; 148:293-300 [ Links ]
68. Iwaki M., Matsuda M., Maeda N., Funahashi T., Marsuzawa Y., Makishima M,. Shimomura I. Induction of adiponectin, a fat derived antidiabetic and antiatherogenic factor by nuclear receptors. Diabetes 2003; 52:1655-1663 [ Links ]
69. Yang, R. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am. J. Physiol. Endocrinol. Metab.2006; 290: E1253-E1261 [ Links ]
70. Hirosumi J. A central role for JNK in obesity and insulin resistance. Nature 2002; 420: 333-336 [ Links ]
71. Shoelson S. Inflammation and the NF- xß axis in obesity- and diet-induced insulin resistance. Int. J. Obes. Relat. Metab. Disord 2003; 27 (Suppl. 3), S49-S52 [ Links ]
72. Howard J. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol. Metab 2006; 17: 365-371 [ Links ]
73. Houstis N, Rosen E. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006; 440: 944-948 [ Links ]
74. Fukuhara A, Matsuda M. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 2005; 307:426 -430 [ Links ]
75. Banerjee R. Regulation of fasted blood glucose by resistin. Science 2004; 303: 1195-1198 [ Links ]
76. Kanda H. MCP-1 contributes to macrophage infiltration in to adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest 2006; 116: 1494-1505 [ Links ]
77. Weisberg S. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest 2006; 116: 115-124 [ Links ]
78. Di Gregorio G. Expresión of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance and reduction by pioglitazone. Diabetes 2005; 54: 2305 -2313 [ Links ]
79. Graham T. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med 2006; 354: 2552-2563 [ Links ]
80. Abizaid A., Gao Q, Horvath T.L. Thoughts for food: brain mechanisms and peripheral energy balance. Neuron 2006; 51: 691 - 702 [ Links ]
81. Martos-Moreno GA, Argente J. Clinical implications of adipokines in pathophysiology: State of the art. J Endorinol Invest 2006 (en prensa) [ Links ]
82. Bjorbaek C, Kahn BB 2004. Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res. 59:305-331 [ Links ]
83. Flier JS Obesity wars: molecular progress confronts and expanding epidemy Cell 2004; 116: 337 -350 [ Links ]
84. Flier JS Clinical review 94: whats in a name in search of leptins physiologic role. Journal of clin Endocrinol Metab 1998; 83: 1407 -1413 [ Links ]
85. Bartness T.J., Banshad M. Innervation of mammalian white adipose tissue: Implications for the regulation of total body fat. Am H Physiol. 1998; 275:R1399-R1411. [ Links ]
86. Bartness T.J., Kay Song C., Bowers R.R., Foster M.T. Brain adipose tissue cross talk. Proc Nutr Soc 2005; 64: 53-64 [ Links ]
87. Lowell B. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature 1993; 366: 740-742 [ Links ]
88. Abu-Elheiga L, Matzuk M. M., Abo Hashema K.A., Wakil S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2 . Science 2001: 291: 2613-2616 [ Links ]
89. Prins JB, ORahilly S 1997. Regulation of adipose cell number in man. Clin Sci (Lond) 92: 3-11. [ Links ]
90. Engfeldt P, Arner P. Lipolysis in human adipocytes, effects of cell size, age of regional differences. Horm Metab Res Suppl. 1998 19:26-29 [ Links ]
91. Depres JP, Fong BS, Julián P, Jimenez J, Angel A. Regional variation in HDL Metabolism in human fat cells: effect of cell size. Am J Physiol 1987;252:E654-659 [ Links ]
92. Blüher M, Wilson-Fritch L, Leszyk J, Laustsen PG, Corvera S, Kahn. Role of insulin actions and cell size on protein expresion patterns in adipocytes. J Biol Chem 2004; 279: 31902 - 31909 [ Links ]
93. Skurk C. Alberti-Huber C. Herder. Relationship between Adipocyte Size and Adipokine Expression and Secretion. J. Clin. Endocrinol. Metab., March 1, 2007; 92(3): 1023 - 1033 [ Links ]
94. Wellen K.E, Hotamisligil G.S. Inflammation, stress and diabetes. J. Clin Invest 2005; 115: 1111 -1119 [ Links ]
95. Hotamisligil G.S., Peraldi P, Budavaria A, Ellis R, White MF, Spiegelman BM. IRS-1 mediated inhibition of insulina receptor tyrosin kinase activity in TNF-alfa and obesity-induced insulin resistance. Science 1996; 271: 665-668 [ Links ]
96. Wellen K.E, Hotamisligil G.S. Inflammation, stress and diabetes. J. Clin Invest. 2005, 115: 1111 -1119. [ Links ]
97. Lin, Y., et al. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactiv oxygen species. J. Biol. Chem. 280:4617-4626 [ Links ]
98. Brownlee,M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001; 414: 813-820. [ Links ]
99. Weisberg, S.P., et al Obesity is associated with macrophage accumulation in adipose tissue. J.Clin. Invest. 2003; 112:1796-1808 [ Links ]
100. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest 2003; 112: 1821 - 1830 [ Links ]
101. Lobby P. Changing concepts of atherogenesis. Journal of Internal Med.2000; 247:349-358 . [ Links ]
102. Sierra-Honigmann, M.R., Nath, A.K., Murakami,C., Garcia Cardena,F., Papapetropoulos, A., Sessa, W.C., Madre,L.A., Schechner, J.S., Schwabb, M.B., Polverini,P.J., Flores-Riveros, J.R. Biological action of leptin as an angiogenic factor. Science 1998; 281: 1683-1686. [ Links ]
103. Bodary, P.F., Westrick,R.J., Wickenheiser, K.J., Shen, Y, Eitzman D.T. Effect of leptin on arterial trombosis following vascular injury in mice. Journal American Med Assoc. 2002; 287: 1706 - 1709 . [ Links ]
104. Xu F.P., Chen M.S., Wang Y.Z., Yi Q, Lin S.B., Chen A.F., Luo J.D. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation 2004; 110: 1269-1275 [ Links ]
105. Ridker P.M., Rifai N., Rose L., Buring J.E.,Cook N.R. Comparison of C reactive protein and low density lipoprotein colesterol levels in the prediction of first cardiovascular events. N Engl Journ Med 2002; 347: 1557-1565 . [ Links ]
106. Stentz F.B., Umpierrez G.E., Cuervo R., Kitabchi A.E., Proinflamatory cytokines, markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes 52(8): 209-2016 [ Links ]
107. Sobel B.E. Increased plasminogen activator inhibitor-1 and vasculopathy. Circulation 1999; 99:2496-2498. [ Links ]
108. Thogersen A.M., Jansson J.H., Boman K. High plasminogen activator inhibitor and tissue plasminogen activator levels in plasma precede a first acute myocardial infarction in both men and women: evidence for the fibrinolytic system as an independent primary risk factor. Circulation 1998; 98: 2241 - 2247 [ Links ]
109. Calabro P., Samudio I., Willerson J.T.,Yeh E. T., Resistin promotes smooth muscle cell proliferation through activation of extracellular signal regulated kinase ½ and phosphatidylinositol 3 kinase pathways. Circulation 2004;110: 3335-334 . [ Links ]
110. Pinkney J.H., Stehouwer C.D., Coppack S.W., Yudkin J.S. Endotelial dysfunction: cause of the insulin resistance syndrom. Diabetes 1997;46:S9-S13. [ Links ]
111. Van Royen N. Hoefer I, Buschmann I, Kostin S, Voskuil M, Bode Ch. Scharper. Effects of local MCP-1 protein therapy on the development of the collateral circulation and atherosclerosis in Watanabe hyperlipidemic rabits. Cardiovasc Res 2003; 57: 178 - 185 . [ Links ]
112. Tan K.C., Xu A, Chow W.S., Lam M.C., Ai V.H., Tam S.C., Lam K.S. Hypoadiponectinemia is associated with impaired endothelium-dependent vasodilation. Journal of Clin Endocrinol Metab 2004; 89:765-769 . [ Links ]
113. Schulze M.B., Dshai I, Rimm E.B., Li T., Rifai N, Hu F.B. Adiponectin and future coronary Heart disease events among men with type 2 diabetes. Diabetes. 2005; 54: 534-539 [ Links ]
114. Lindsay R.S., Resnick H.E., Zhu J., Tun M.L., Howard B.V., Zhang Y., Yeh J., Best L.G. Adiponectin and coronary heart disease: the strong heart disease. Arterioscler Thromb Vasc Biol. 2005 Mar; 25(3): 15-6. [ Links ]
115. Makino T., Noguchi Y., Yoshikawa T., Doi C., Nomura K., Circulating interleukin 6 concentrations and insulin resistance in patients with cancer. British journal of Surgery 1998; 85: 1658 -1662. [ Links ]
116. Flier JS, Harris M, Hollenberg AN 2000. Leptin, nutrition and the thyroid: the why, the wherefore, and the wiring. J Clin Invest 105:859-861. [ Links ]
117. Chan JL, Heist K, DePaoli A, Veldhuis JD, Mantzoreos CS 2003. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short term starvation in healthy men. J Clin Invest 111:1409-1421. [ Links ]
118. Margetic S, Gazzola C, Pegg GG, Hill RA 2002. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord 26:1407-1433. [ Links ]
119. Lord GM, Materese G, Howard JK, Baker RJ, Bloom SR, Lechler RI 1998. Leptin modulates the T-cell immune response and reverses starvation-induced immunossuppresion. Nature 394:897-901. [ Links ]
120. Cock TA, Auwerx J 2003 Leptin: cutting the fat off the bone. Lancet 362:1572-1574 [ Links ]
121. Klein J., Perwitz N., Graus D., Fasshauer M. Adipose tissue as a source and target for novel therapies. Trends in endocrinology and metabolism. 2006; 17(1): 26 -32 [ Links ]
122. Goossens GH, Blaak EE, van Baak MA 2003. Possible involvement of adipose tissue renin angiotensin system in the pathophisology of obesity and obesity related disorder Obes Rev 4:43 -55. [ Links ]
123. Pollare Tm Lithell H, Berne C 1989 A comparison of the effects of hydrocholorothiazide and captopril on glucose and lipid metabolism in patient with hypertension. N Engl J Med 321:868-873 [ Links ]
124. Petrie JR, MOrris AD, Ueda S, Samll M, Donnelly R, Connel JM, Elliott HL 2000 Trandolapril does not improve insulin sensitivity in patients with hypertension and type 2 diabetes : a double-blind, placebo-controlled crossover trial J Clin Endocrinol Metab 85:1882-1889. [ Links ]
125. Di Gregorio, G. B. et al. Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes 2005 54, 2305-2313. [ Links ]

