










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Med
Print version ISSN 0121-5256On-line version ISSN 1909-7700
rev.fac.med vol.16 no.1 Bogotá Jan./June 2008
PRÁCTICA CLÍNICA
ENFERMEDAD DE ORINA EN JARABE DE ARCE: MEJORIA CLÍNICA ASOCIADA A DETECCIÓN PRECOZ Y MANEJO OPORTUNO. REPORTE DE CASO Y REVISIÓN DE LITERATURA
MAPLE SYRUP URINARY DISEASE: CLINICAL IMPROVEMENT ASSOCIATED WITH EARLY DETECTION AND MANAGEMENT. CASE REPORT AND LITERATURE REVIEW
DOENÇA DE URINA EM XAROPE DE ARCE: MELHORIA CLÍNICA RELACIONADA Á DETECÇÃO PRECOCE E MANEJO OPORTUNO. REPORTE DE CASO E REVISÃO DE LITERATURA
JUAN FERNANDO GÓMEZ-CASTROa*, EUGENIA ESPINOSA-GARCÍAa, b, LUIS ALEJANDRO BARRERAc Y OLGA Y. ECHEVERRYc
a Programa Neurología Pediátrica, Universidad Militar Nueva Granada, Bogotá D.C., Colombia.
b Servicio de Neuropediatría, Hospital Militar Central, Bogotá D.C., Colombia.
c Instituto de Errores Innatos del Metabolismo, Pontificia Universidad Javeriana, Bogotá D.C., Colombia.
* Correspondencia: jfgomezcastro@gmail.com. Dirección Postal: Servicio de Neuropediatría, Hospital Militar Central, Tr. 3 No. 49-00, Bogotá D.C., Colombia.
Recibido: Enero 21 de 2008. Aceptado: Febrero 13 de 2007.
Resumen
La enfermedad de orina en jarabe de arce es un error innato del metabolismo de los cetoácidos de cadena ramificada, cuya acumulación produce una encefalopatía neonatal grave y que de no ser diagnosticada y tratada de forma precoz y oportuna, lleva invariablemente a la aparición de secuelas neurológicas permanentes y a un posterior desenlace letal. El presente articulo busca, mediante la descripción de un caso clínico sucedido en el Hospital Militar Central de Bogotá, hacer una revisión de la literatura acerca de la enfermedad, resaltando los mecanismos fisiopatológicos, la detección por diferentes pruebas de laboratorio, así como las estrategias de manejo, demostrando que gracias a los progresos realizados en su comprensión y enfoque, actualmente se puede hablar de evitar la mortalidad, alcanzando en muchos casos, una sobrevida a largo plazo sin mayores secuelas neurológicas, todo ello con un manejo interdisciplinario que logre un control metabólico adecuado.
Palabras clave: Errores innatos del metabolismo, cetoácidos.
Abstract
Maple syrup urine disease is an inborn error of the metabolism of branched chain keto-acids whose accumulation produces a serious neonatal encephalopathy, which if not diagnosed and treated in a precocious and opportune way, will invariably lead to the appearance of permanent neurological impairments and an ulterior lethal outcome. The present article intends, by means of the description of a clinical case which occurred at the Hospital Militar Central, to perform a review of the existent literature on this disease, to revise its fisiopathological mechanisms as well as its detection using different laboratory tests and the different care strategies, to demonstrate that, thanks to the progress achieved in its understanding and focus, at the present moment we can speak of avoiding mortality, accomplishing in many cases long term survival without important neurological consequences, all by means of an interdisciplinary approach that achieves an appropriate metabolic control.
Key words: Metabolism inborn errors, keto acids.
Resumo
A doença de urina em xarope de arce é um erro inato do metabolismo dos cetoácidos de corrente ramificada, cuja acumulação produz uma encefalopatia neonatal grave e que de não ser diagnosticada e tratada de forma precoce e oportuna, leva invariavelmente à aparição de seqüelas neurológicas permanentes e a um posterior desenlace letal. O presente artigo procura, mediante a descrição de um caso clínico sucedido no Hospital Militar Central de Bogotá, fazer uma revisão da literatura a respeito da doença, ressaltando os mecanismos fisiopatológicos, a detecção por diferentes provas de laboratório, bem como as estratégias de tratamento, demonstrando que graças aos progressos realizados em seu entendimento e enfoque, atualmente se pode falar de evitar a mortalidade, atingindo em muitos casos, uma sobrevida em longo prazo sem maiores seqüelas neurológicas, tudo isso com um manejo interdisciplinares que consiga um controle metabólico adequado.
Palavras-chave: Erros inatos do metabolismo , cetoácidos.
Reporte de caso
Menor femenino producto de primer embarazo mediante inseminación artificial de madre de 30 años de edad. No consanguíneos, embarazo de curso normal, parto vaginal a las 39 semanas con adecuada transición a la vida extrauterina, llanto y respiración espontánea. Peso al nacer de 2734 g, talla de 47cm y APGAR 9-10-10. Egresó sin complicaciones y reingresó un día después, al notar los padres somnolencia y pobre succión. El examen físico confirmó lo anterior y presencia de hipotono generalizado; no había abombamiento de suturas, diastasis de suturas u otros signos de hipertensión endocraneana y la orina no presentó algún olor característico. Se inició hidratación IV y se realizaron paraclínicos con lo siguientes resultados:


Con los resultados anteriores se realizó cromatografía de aminoácidos (AA) que demostró elevaciones anormales de valina, leucina e isoleucina; se continuó con el mismo manejo, se realizó una resonancia cerebral que evidenció hiperintensidades inespecíficas del tallo cerebral y de los ganglios basales y una cromatografía de ácidos orgánicos que demostró elevaciones de los ácidos cetoisocaproíco, cetovalérico e hidroximetilvalérico. Los resultados confirmaron el diagnóstico de enfermedad de orina en jarabe de arce, se continuó con el manejo, se reinició vía oral con restricción proteica y se adelantó consecución de fórmula de alimentación especial libre de AA de cadena ramificada. Se documentó mejoría del estado general, reinicio de succión con adecuada tolerancia oral, mejoría del estado de conciencia con períodos de sueño y vigilia y mejoría del tono muscular. Tras la consecución de fórmula de alimentación especial se egresó con el manejo instaurado. A la fecha de elaboración de este artículo, enero de 2008, la niña cuenta con siete meses de vida y continúa con manejo sin presentar nuevos eventos de descompensación metabólica. Los controles de cromatografía de AA son normales, presentando una leve disminución del tono postural con desarrollo neurológico normal. El manejo actual incluye una combinación monitorizada de fórmula láctea maternizada, fórmula especial libre de aminoácidos ramificados (MSUD analogg), carnitina, benzoato de sodio en descenso gradual y suplemento de tiamina 50 mg por día.
Definición de la enfermedad
La enfermedad de orina en jarabe de arce (MSUD por sus siglas en inglés Maple Syrup Urine Disease), es un error innato del metabolismo caracterizado por acumulación de AA de cadena ramificada y de alfa cetoacidos ramificados. Fue reportada por primera vez en 1954 por Menkes (1) quien detectó tres casos en una familia y se describe como "enfermedad neurodegenerativa" de inicio neonatal y con desenlace letal hacia el tercer mes de vida; los afectados presentan un olor particular en la orina, semejante al jarabe de arce usado como alimento.
El reconocimiento del defecto enzimático responsable de la MSUD se inicia con la documentación de niveles plasmáticos elevados de AA de cadena ramificada, valina, leucina e isoleucina (BCAA), en los individuos afectados (2), para posteriormente aislarse también los alfa cetoácidos ramificados (BCKA), sugiriendo que el defecto metabólico se encuentra en el catabolismo de AA, particularmente en la descarboxilación de cetoácidos (3). Investigaciones posteriores han confirmado la presencia del defecto en leucocitos y han llevado al aislamiento y a la homogenización del complejo enzimático de deshidrogenasas para cetoácidos ramificados, así como de las quinasas y fosfatasas específicas (4-5).
La MSUD es reconocida como una enfermedad hereditaria autosómica recesiva de distribución multiétnica, con una frecuencia estimada de 1:185.000 (6); el reconocimiento de variantes fenotípicas más benignas de la enfermedad indicaría que existe un subregistro de los pacientes afectados. Hay comunidades con mayor incidencia de MSUD, dentro de ellas la mennonita norteamericana y algunas del Oriente Medio. Por técnicas de genética molecular se ha logrado la clonación del ADN para subunidades catalíticas en los afectados, identificando así la localización cromosómica de las tres subunidades del complejo enzimático y la estructura de sus tres genes (7). En la actualidad se descubren cada vez más mutaciones para los loci de las subunidades implicadas en MSUD, lo que se traduce en una mejor identificación prenatal, en el reconocimiento de los portadores y en la redistribución en los diferentes grupos de población (8).
Fisiopatología
La leucina, la isoleucina y la valina son AA esenciales de cadena ramificada y constituyen la mitad de los requerimientos en la dieta de animales superiores; sus funciones biológicas comprenden la síntesis proteica, la de ácidos grasos y colesterol, la restauración del balance nitrogenado y un freno del catabolismo que se observa en condiciones como sepsis y politraumatismo (9). Su metabolismo incluye incorporación a proteínas y procesos de degradación oxidativa en las mitocondrias, siendo importantes en el músculo como fuente alterna de energía, así como en el cerebro, en los riñones, en el corazón y en los tejidos grasos. Los ácidos a-cetoisocaproico, a-cetometilisovalérico y a-cetoisovalérico derivados de la leucina, de la isoleucina y de la valina, son alfa cetoácidos ramificados que se metabolizan en el hígado para producir cuerpos cetónicos y derivados coenzima A (CoA); en la figura 1 se representan las vías para su degradación fisiológica.
Tras ingresar a la célula a través de un transportador L independiente de sodio, los BCAA realizan tres pasos: inicialmente se presenta descarbamilación citosólica o mitocondrial, que da como producto BCKA; enseguida transcurre proceso de descarboxilación oxidativa mitocondrial catalizado por un complejo enzimático de BCKA-deshidrogenasas, que da como resultado subproductos derivados de CoA de cadena ramificada y en un tercer paso, son metabolizados mediante deshidrogenación, lo que da como productos finales succinil CoA, acetoacetato y Acetil CoA, que luego ingresan a otros procesos metabólicos como el ciclo de Krebs y la gluconeogénesis (10).
El defecto principal de MSUD reside en la deficiente actividad del complejo enzimático de deshidrogenasas para los cetoácidos de cadena ramificada, lo que implica que los pasos metabólicos a partir de este se verían suprimidos, con acumulación de los subproductos generados al paso que se considera "bloqueado" (Figura 2).
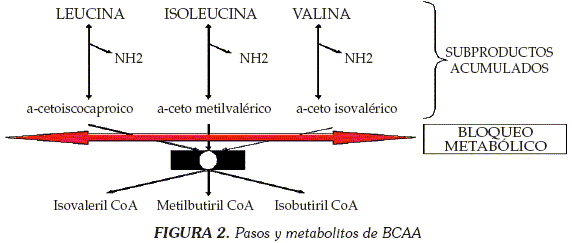
El efecto principal de los metabolitos acumulados se produce en el cerebro, ocasionando un efecto tóxico directo (11) que se traduce en disfunción y en eventualmente muerte de las neuronas. Por una combinación de edema citotóxico y de edema intersticial producido por la liberación de mediadores inflamatorios, se genera edema cerebral, responsable a su vez de abombamiento de fontanelas y de diastasis de suturas, que precipitan a un desenlace fatal de la enfermedad (12).
Manifestaciones clínicas y pruebas de laboratorio
El cuadro clínico corresponde al de una encefalopatía aguda durante los primeros días de la vida neonatal (13), aunque hay formas más leves que pueden tener su presentación inicial en edades mayores. La combinación de niveles normales de amonio, en ausencia de acidosis metabólica (observada en estados iniciales de la enfermedad) en un neonato en coma, o con signos de encefalopatía, deben aumentar la sospecha de la enfermedad (figura 3).
Como los síntomas de la enfermedad sólo son aparentes al estar instaurada la encefalopatía, se trata de por si de una situación grave que compromete las posibilidades de recuperación, a pesar de un manejo adecuado (15); de ahí que la detección precoz se convierta en un factor determinante para evitar secuelas neurológicas irreversibles y para reducir la mortalidad. Las manifestaciones iniciales de MSUD aparecen generalmente a partir del cuarto día de vida, siendo la letargia y la succión pobre los signos más precoces; a partir del séptimo día se empieza a advertir pérdida de peso y cambios en el tono muscular, siendo el momento en el que también aparece un olor característico en la orina, semejante al azúcar quemada y secundario al acúmulo de isoleucina y de 4-5-dimeti-3-hidroxifuranona que se evidencia por la presencia de cetonas. Si la enfermedad progresa sin tratamiento los signos de hipertensión endocraneana aparecen al final de la segunda semana de vida, con progresión del deterioro de conciencia hasta llegar al coma y finalmente a la muerte.
Los exámenes paraclínicos permiten descartar otras causas potenciales de encefalopatía neonatal, sea esta metabólica o no. La detección de cetoácidos en orina (prueba de dinitrofenilhidrazina) no es exclusiva de MSUD, por lo que los niveles elevados de los BCAA mediante la cromatografía de AA, o por cromatografía de ácidos orgánicos, así como la detección de metabolitos intermedios que se encuentren elevados o disminuidos, de acuerdo a la evolución de la enfermedad, se necesitan para confirmar el diagnóstico (16).
Conocido el rol de la leucina y de su cetoácido como responsables del daño en la enfermedad, su detección es de gran importancia en el diagnóstico y seguimiento. Se conoce que tras la restricción proteica y la rápida hidratación se aumenta la excreción de metabolitos tóxicos, con reducción de sus niveles a las primeras 24 horas de instaurado el tratamiento, hasta la normalización de sus valores (17). Es por ello que el seguimiento a largo plazo cuenta con esta prueba para la detección precoz de una descompensación aguda y para el monitoreo del tratamiento; no se puede hablar de estandarización respecto a la periodicidad con que se deba realizar, quedando entonces esta decisión a criterio del clínico. Los inconvenientes que implican la poca disponibilidad de dichas pruebas en la mayoría de centros asistenciales llevan a demostrar que su reducción sigue un modelo cinético biexponencial, similar al manejado por ciertas moléculas pequeñas como la urea, proponiéndose entonces un patrón predecible de reducción de la leucina cuando el paciente está en manejo para desintoxicación y de esta manera disminuir la necesidad de seguimientos periódicos a través de cromatografías de ácidos orgánicos(18).
Clasificación
Según la forma de presentación y del patrón de respuesta clínica a la tiamina, administrada como parte del manejo, la MSUD se puede clasificar en cinco fenotipos (Tabla 1), clínicamente distinguibles los unos de los otros (15). Sin embargo, la clasificación de un paciente en un fenotipo o en otro se puede llegar a dificultar, por la falta de criterios para distinguirlas. Para el tipo clásico hay características que permiten reconocerla, como la rapidez en la aparición y la severidad del cuadro. Aún así, cualquier MSUD que aparezca durante el período neonatal se debería considerar como variante clásica, hasta que su evolución demuestre lo contrario. La variante intermedia, considerada de inicio más tardío, cursa con cetoacidosis y raramente presenta descompensación aguda que lleve a encefalopatía (19). Las formas intermitentes cursan con neurodesarrollo más cognición normal y tienen riesgo de descompensación aguda letal, ante eventos que induzcan estrés metabólico o catabolismo (20). Las formas respondedoras a tiamina se caracterizan por respuestas favorables y sostenidas a la restricción dietaria y a administración del cofactor en dosis entre 10 a 1000 mg/día; su evolución durante el manejo es variable, mencionándose casos de esta variante que llegan a cursar con cognición normal y con desempeño social adecuado en la vida adulta (21). Los afectados con la variante de deficiencia en el complejo dihidrolipoil deshidrogenada (E3), una rara forma de la enfermedad, cursan con un cuadro similar a MSUD, pero acompañado de acidosis láctica que progresa hasta hacerse grave en los primeros meses de vida (22).
Tratamiento
Con el fin de minimizar la acumulación de intermediarios lesivos, el manejo de las deficiencias en el metabolismo de los AA esenciales requiere de limitar su consumo, hasta llegar al mínimo requerido para satisfacer las necesidades que implican el crecimiento y desarrollo del afectad (15). Está demostrado que el buen control metabólico se correlaciona con un mejor resultado intelectual a largo plazo en los pacientes con MSUD.
El manejo de esta enfermedad se puede dividir en dos fases, el manejo agudo y el manejo de sostén. En la descompensación aguda (manejo agudo) se debe ser agresivo y se comprenden tres puntos:
a) Remoción rápida del tóxico: La hemodiálisis, la diálisis peritoneal o ambas, tienen mayor efectividad en el aclaramiento de BCAA y BCKA (23); las dificultades técnicas de este procedimiento en lactantes son factores que limitan el uso en estos pacientes. El uso de medicamentos para fomentar la producción de ésteres excretables o metabolizables ha sido asumido como terapéutica eficaz para el control de los errores innatos del metabolismo, considerando que ante la menor señal de pobre respuesta al manejo, se debe acompañar de otra terapéutica que permita detoxificar rápida y efectivamente al paciente. La tiamina ha demostrado efectividad en las formas respondedoras, por lo que todo paciente con MSUD de reciente diagnóstico se debería someter a un ensayo clínico de tres semanas para evaluar si corresponde a las formas sensibles a dicho suplemento.
b) Soporte nutricional: Se han ensayado diversas terapias de alimentación parenteral y su uso se debe realizar en combinación con otras terapéuticas, demostrándose efectividad en el manejo de formas leves de descompensación (24). Teniendo en cuenta que el descenso de las concentraciones plasmáticas de isoleucina y de valina es más rápido que el de la leucina, se necesita reiniciar su aporte tras uno o dos días de haber iniciado el tratamiento.
c) Manejo catabolismo / anabolismo: Durante estadios de infección, de ayuno o de vacunación, se aumenta el catabolismo proteico y se da un aumento endógeno de BCAA (25). La reducción del aporte proteico dietario y la instauración de fórmulas especiales previene el deterioro de la descompensación aguda y en pacientes con adecuado control metabólico no se ha demostrado que la vacunación precipite las crisis de la enfermedad (16).
En el manejo de sostén el tratamiento consiste en normalizar niveles de BCAA limitando la ingesta de los mismos, mientras que se provee una nutrición que mantenga el desarrollo y crecimiento. Las formulas comerciales disponibles se basan en una dieta libre de BCAA que brinden un aporte de 2 a 3 g/K/día de equivalente proteico y de 20 a 24 Kcal/oz de aporte calórico. Los controles se deben realizar midiendo semanalmente los BCAA durante la fase inicial, para luego hacerlos menos frecuentes. También se dispone de sistemas de monitoreo urinario para el control de la enfermedad (16).
Pronóstico
Las mejoras en el entendimiento, la monitoria y el manejo oportuno y adecuado de los pacientes permiten disminuir la morbilidad y la mortalidad por MSUD. La edad de diagnóstico y el curso de la enfermedad siguen siendo factores independientes, que pueden afectar el pronóstico a largo plazo. El riesgo de presentar complicaciones por descompensación aguda sigue siendo elevado hasta los seis años de vida, tiempo en el que el organismo es capaz de tolerar mejor el estrés. Se ha observado que los pacientes que presentan pobre desarrollo intelectual en general, ya cursaban con otras secuelas neurológicas (28). Las perspectivas para el futuro incluyen la instauración del tamizaje neonatal general para ésta y para otras patologías metabólicas que requieren de un diagnóstico precoz como forma de instaurar manejos oportunos (29).
Referencias
1. Menkes JH. A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urine substance. Pediatrics. 1954;14:462-63. [ Links ]
2. Westall RG. Maple syrup disease. Isolation and identification of organic acids in the urine. Pediatrics. 1959;23:348-49. [ Links ]
3. Dancis J. Maple syrup urine disease: branched-chain keto-aciduria. Pediatrics. 1960;25:72-3. [ Links ]
4. Pettit FH. Purification and characterization of branched chain keto-acid dehydrogenase complex of bovine kidney. Proc Natl Acad Scie USA. 1978;75:4881-82. [ Links ]
5. Damuni Z. Purification and properties of the catalytic subunit of the branched chain keto-acid dehydrogenase phosphatase from bovine kidney mitochondria. J Biol Chem. 1987;262:5129-30. [ Links ]
6. Naylor EW. Newborn screening for maple syrup urine disease. En: Bickel H, editor. Neonatal screening for inborn errors of metabolism. Berlin: Springer-Verlag; 1980. p. 19-28. [ Links ]
7. Mitsubushi H. Structural organization and chromosomal localization of the gene for the E1B subunit of human branched chain keto-acid dehydrogenase. J Biol Chem. 1991;266:1466-67. [ Links ]
8. Chuang D. Disorders of branched chain aminoacid and keto acid metabolism. En: Scriver C, editor. The metabolic and molecular bases of inherited disease. New York: Mc Graw-Hill; 2001. p. 1239-40. [ Links ]
9. Freund HR. The use of branched-chain amino acids in the injured-septic patient. En: Wasler M, editor. Metabolism and clinical implication of branched chain amino and ketoacids. New York: Elsevier; 1981. p. 527-28. [ Links ]
10. Harper AE. Branched-chain aminoacid metabolism. Annu Rev Nutr. 1984;4:409-10. [ Links ]
11. Breningstall G. Approach to diagnosis of oxidative metabolism disorders. Ped Neurol. 1993;2:253-54. [ Links ]
12. Treacy E. Interrelations between branched-chain amino and hydroacids, implications for treatment, associations with CNS dysmyelination. J Inherited Metab Dis. 1992;15:121-22. [ Links ]
13. Lyon G. Neurology of hereditary metabolic diseases in children. 2 nd ed. New York: Mc Graw-Hill; 1996. [ Links ]
14. Hoffman G. Inherited metabolic diseases: a clinical approach. 2 nd ed. Philadelphia: Lippincott William & Wilkins; 2004. [ Links ]
15. Peinemann F. Maple syrup urine disease. J Inherit Metab Dis. 1994;17:3-4. [ Links ]
16. Chuang D. Disorders of branched chain aminoacid and keto acid metabolism. En: Scriver C, editor. The metabolic and molecular bases of inherited disease. New York: Mc Graw-Hill; 2001. p. 1239-40. [ Links ]
17. Hmiel S. Amino acid clearance during acute decompensation in maple syrup urine disease treated with hemofiltration. Pediatr Crit Care Med. 2004;5(3):278-79. [ Links ]
18. Jouvet P. Kinetic modeling of plasma leucine level during continuous venous extracorporeal removal therapy in neonates with maple syrup urine disease. Pediatr Res. 2005;58(2):278-79. [ Links ]
19. Muller H. MSUD with an intermittent relatively benign course. Verlauf Dtsch Med Wochenschr. 1971;96:1552-53. [ Links ]
20. Dancis J. Intermittent branched-chain ketonuria, variant of MSUD. N Eng J Med. 1967;276:84-5. [ Links ]
21. Riviello J. Cerebral edema causing death in children with MSUD. J Pediatr. 1991;119:42-3. [ Links ]
22. Harper PA. MSUD as a cause of spongiform encephalopathy in calves. Vet Rec. 1986;119:62-3. [ Links ]
23. Rutledge SL. Neonatal hemodialysis: effective therapy for the encephalopathy of inborn errors of metabolism. J Pediatr. 1990;116:125-26. [ Links ]
24. Berry GT. Branched-chain amino acid-free parenteral nutrition in the treatment of acute decompensation in patients with MSUD. N Eng J Med. 1991;324:175-76. [ Links ]
25. Thompson GN. Acute illness in MSUD: dynamics on protein metabolism and implications for management. J Pediatr. 1991;119:35-6. [ Links ]
26. Snyderman SE. Treatment outcome of maple syrup urine disease. Acta Paediatr Jpn. 1988;30:417-18. [ Links ]
27. Kaplan P. Intellectual outcome in children with MSUD. J Pediatr. 1991;119:46-7. [ Links ]
28. Naughten ER. Outcome of maple syrup urine disease. Arch Dis Child. 1982;57:918-19. [ Links ]
29. Hiliges C. The intellectual performance of patients with MSUD. Eur J Pediatr. 1993;152(2):123-9. [ Links ]

