










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Med
Print version ISSN 0121-5256On-line version ISSN 1909-7700
rev.fac.med vol.16 no.2 Bogotá July/Dec. 2008
PRÁCTICA CLÍNICA
COMPROMISO SISTÉMICO POR LINFOMA INTRAVASCULAR DE CÉLULA GRANDE
SYSTEMIC COMPROMISE FROM LARGE CELL INTRAVASCULAR LYMPHOMA
COMPROMISSO SISTÉMICO POR LINFOMA INTRAVASCULAR DE CÉLULA GRANDE
JOSÉ J. ARIAS A. M.D.a*, LAURA ESCOBAR M.D.a Y MARÍA HELENA ASTORQUIZA, M.D.a
a Servicio de Patología, Hospital Militar Central, Bogotá, D.C., Colombia, Universidad Militar Nueva Granada, Bogotá D.C., Colombia.
* Correspondencia: jjariasa@yahoo.com. Dirección postal: Bogotá, D.C.
Recibido: Noviembre 7 de 2007. Aceptado: Febrero 20 de 2008.
Resumen
Se presenta el primer caso de linfoma intravascular de célula grande, una variante del linfoma de células B grandes difuso, diagnosticado en el Hospital Militar Central de Bogotá, en una mujer adulta mayor, quien consultó por un síndrome febril prolongado, pérdida de peso, síntomas constitucionales y quien veinte días antes del ingreso perdió fuerza progresiva, hasta la paraplejia. Se iniciaron estudios para descartar enfermedades neoplásicas, infecciosas y autoinmunes, pero falleció el día catorce de su hospitalización. El estudio post-mortem, realizado cinco horas después, mostró una lesión tumoral maligna de célula grande, limitada a los espacios vasculares de múltiples vísceras. Se planteó como diagnóstico una neoplasia de origen linfoide, que se confirmó posteriormente mediante pruebas inmunohistoquímicas.
Palabras clave: linfoma de células B grandes difuso, linfoma de células B
Abstract
This is the first case of intravascular large cell lymphoma diagnosed at the Hospital Militar de Bogota, in an adult female who presented with chronic fever, weight loss, constitutional symptoms and progressive motor deterioration for the previous 20 days to the point of paraplegia. A work up was carried out to rule out neoplastic diseases, infections or autoimmune diseases, but she died on the fourteen hospital day. The autopsy, performed 5 hours post mortem, showed an intravascular large cell malignancy, limited to the vascular spaces of multiple organs. The lymphomatous origin was confirmed posteriorly by immunochemical tests.
Key words: lymphoma large B-cell diffuse, lymphoma B-cell
Resumo
Apresenta-se o primeiro caso de linfoma intravascular de célula grande, uma variante do linfoma de células B grandes difuso, diagnosticado no Hospital Militar Central de Bogotá, numa mulher adulta maior, quem conferiu por uma síndrome febril prolongado, perda de peso, sintomas constitucionais e quem vinte dias antes do rendimento perdeu força progressiva, até a paraplegia. Iniciaram-se estudos para descartar doenças neoplásicas, infecciosas e auto-imunes, mas faleceu o dia quatorze de sua hospitalização. O estudo post-mortem, realizado cinco horas depois, mostrou uma lesão tumoral maligna de célula grande, limitada aos espaços vasculares de múltiples vísceras. Propôs-se como diagnóstico uma neoplasia de origem linfóide, que se confirmou posteriormente mediante provas inmunohistoquímicas.
Palavras-chave: Linfoma difuso de grandes células, linfoma de células B
Introducción
Los linfomas corresponden a la cuarta o quinta causa de tumores malignos y la mayoría de ellos se originan en linfocitos B. En la población adulta los linfomas foliculares y el linfoma difuso de célula B corresponden a la mayoría de las neoplasias diagnosticadas, mientras que la enfermedad de Hodgkin (linfoma de Hodgkin) representa un porcentaje menor de los casos. El linfoma intravascular de célula grande es una variante inusual del linfoma difuso de célula grande originado generalmente en linfocitos B y su diagnóstico se puede confirmar empleando técnicas de inmunohistoquímica. El propósito de este artículo es el de presentar el primer caso de linfoma de célula grande intravascular diagnosticado en el Hospital Militar Central, confirmado en material de autopsia con la técnica de inmunoperoxidasa.
Presentación del caso
Paciente femenina de 64 años con cuadro clínico de un año de evolución, consistente en síntomas respiratorios altos en forma frecuente, fiebre elevada cuantificada en 39°C de predominio vesperal, malestar general, sensación de distensión abdominal y dolor, astenia, adinamia, hiporexia perdida de peso, diaforesis -principalmente nocturna- de 20 días de evolución, progresiva pérdida de fuerza en miembros inferiores hasta la paraplejia y perdida de la sensibilidad asociada. Permaneció hospitalizada durante un mes en una clínica fuera de Bogotá donde se le realizaron múltiples exámenes y al no encontrarle un diagnóstico definitivo se decidió su traslado al Hospital Militar Central de Bogotá para estudio y manejo.
Revisión por sistemas: Pérdida de peso no cuantificada durante el último año.
Antecedentes quirúrgicos: Amigdalectomia, resección de ganglión en mano derecha, resección de masa quística en seno derecho, negando otros antecedentes.
Examen físico de ingreso: Paciente en mal estado general, somnolienta, hipotensa, taquicárdica, afebril, con onda ascítica y edema de pared abdominal y edema grado III de miembros inferiores. Al examen neurológico se encuentró orientada en las tres esferas afecto lábil, memoria reciente normal, evocativa comprometida, no calculaba déficit de abstracción y juicio, fondo de ojo normal, fuerza muscular marcadamente disminuida en miembros inferiores (de 0-1/5), reflejo aquiliano abolido, reflejo rotuliano presente levemente de forma bilateral, nivel sensitivo T9, atonía de esfínter anal, compromiso de sensibilidad superficial y marcha profunda no valorable.
Exámenes paraclínicos: trombocitopenia persistente con rangos entre 5.000 y 80.00 plaquetas por mm3, anemia leve, sin alteraciones en las pruebas de función renal y hepática.
Imágenes diagnósticas: En la hospitalización inicial se realizó ecografía abdominal que mostró ascitis severa sin visceromegalias, TAC abdominal donde se observó leve aumento de tamaño del riñón derecho y endoscopia de vías digestivas que reveló gastritis superficial corporoantral.
Evolución: Al considerarse como paciente con cuadro febril prolongado de origen desconocido, se intentó descartar proceso paraneoplásico vs tuberculosis y posible insuficiencia suprarrenal. Se hospitalizó, se inició reanimación y manejo para el estado de choque inicial, se transfundió con tres unidades de glóbulos rojos y se inició tratamiento con hidrocortisona por sospecha de falla adrenal. Los estudios infecciosos para tuberculosis, hemoparásitos y leptospirosis fueron negativos, así como los realizados para descartar enfermedades autoinmunes. Fue valorada por el Servicio de Neurología ordenándole estudio de potenciales evocados y de electroconducción con velocidades de conducción de nervios normales amplitud baja, electromiografía y estudio de neuropatía axonal sensitivo motora crónica. Tras la sospecha de TBC diseminada se inició manejo antituberculoso. Se evidenció aumento de las bilirrubinas totales (6,7 mg/dl) a expensas de la directa (5,2 mg/dl) con un aumento muy leve de las aminotrasnferasas. Por el cuadro anémico y la trombocitopenia importantes y persistentes se le transfundieron nuevamente glóbulos rojos y plaquetas y se realizó mielograma que reveló médula reactiva con hiperplasia eritroide, anisocitosis ligera con microcitosis, algunas macroplaquetas y trombocitopenia. La paciente continuó con evolución tórpida, unos días con mejoría clínica importante y otros con deterioro y trombocitopenia persistente que oscilaba entre 10.000 y 5.000 plaquetas por mm3.
Se consideró una probable causa paraneoplásica, por lo que se le descartaron tumores de origen ginecológico, se le realizó una resonancia nuclear magnética de columna dorso lumbar que evidenció hiperseñal intramedular de naturaleza reactiva en T2, extendida al cuerpo vertebral de L1 sin efecto expansivo. Las pruebas para complemento (C3 y C4) y ANAS fueron negativas, c- ANCA 165 (positivo) y p-ANCA 4,7 (negativo). Dado el cuadro clínico con compromiso neurológico, los hallazgos imaginológicos y las pruebas inmunológicas, se consideró una probable poliarteritis nodosa, sin embargo la paciente sufrió un súbito deterioro del estado de conciencia con alteración del patrón respiratorio, desviación de la mirada a la derecha, hipotensión marcada, taquicardia, reflejos pupilares ausentes con isocoria y movimientos de descerebración bilateral. Dado el cuadro de trombocitopenia y deterioro extremos, se sospechó de un accidente cerebro vascular hemorrágico, por lo que se decidió iniciar intubación orotraqueal, se aumentaron inotrópicos pero la paciente falleció el día catorce de su hospitalización con los siguientes posibles diagnósticos: Tuberculosis diseminada; Poliarteritis nodosa; Enfermedad cerebrovascular hemorrágica, Mal de Pott, Síndrome constitucional de desnutrición grave.
Estudio postmortem: Cinco horas después del fallecimiento se realizó el estudio postmortem de una mujer adulta mayor, que al examen externo presentaba estado de desnutrición aparente, con múltiples lesiones petequiales y purpúricas en cara anterior de muslos y en hemiabdomen inferior, hematomas en sitios de venopunción en cara anterior de brazos y a nivel cervical yugular izquierdo, distensión abdominal, onda ascítica positiva, edema de pared abdominal y edema grado III de miembros inferiores. Al examen de cavidades presentaba aproximadamente 1.000 cm3 de liquido serohemático en cada hemitórax, aproximadamente 2.000 cm3 en cavidad abdominal y cavidad craneana sin colecciones. Al examen de los órganos el encéfalo pesó 1370 g, se observaron múltiples petequias que comprometían principalmente lóbulos frontoparietales a nivel basal bilateral, así como cerebelo y tronco. No se observaron alteraciones a nivel de los vasos del polígono de Willis, había leve congestión leptomeníngea y la duramadre y los senos venosos no presentaban alteraciones macroscópicas. A los cortes también se observaron áreas de hemorragia intraparenquimatosa que comprometían lóbulos frontal, parietal, temporal y occipital, núcleos basales, cerebelo y tronco cerebral (Figura 1).

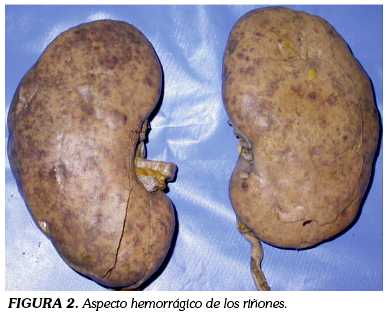
Los riñones tenían peso normal con superficie externa lisa, de color pardo-rojizo y con lesiones petequiales puntiformes tanto externamente como al corte (Figura 2).
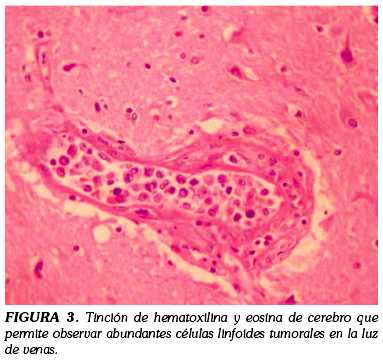
El estudio histológico con hematoxilina eosina mostró una población de células tumorales grandes, con núcleos pleomórficos, vesiculares y nucleolo prominente que ocupaban los espacios vasculares en todas las vísceras examinadas (Figura 3).
Para establecer el origen de las células tumorales se realizaron pruebas con inmunoperoxidasa (Figura 4) que resultaron positivas para los marcadores de linfocitos B (DAKO Laboratories, Carpintería, California, USA).

Se consideró que la muerte ocurrió por hipertensión endocraneana, debido al compromiso tumoral del sistema nervioso central.
Discusión
El linfoma intravascular es un subtipo raro de linfoma de célula grande B, caracterizado por la presencia de células tumorales únicamente en la luz de estructuras las vasculares, sin compromiso ganglionar y sin formación de masas. La enfermedad fue descrita en 1959 por Pfleger y Tappeiner en Alemania y desde su descubrimiento ha recibido diferentes nombres: angioendoteliomatosis sistémica proliferante, angioendoteliomatosis maligna, linfomatosis intravascular, linfoma angioendoteliotrópico (clasificación Kiel), linfoma angiotrópico de célula grande (clasificación de Lucks y Collins), linfoma difuso de célula grande (clasificación de Real, clasificación revisada de linfoma, Europeo-Americana) y linfoma intravascular según la Organización Mundial de la Salud (1). Su origen linfoide se determinó mediante pruebas inmunohistoquímicas en 1986, predominando los de origen B y en muy pocos caso los de origen T, histiocítico y NK (2).
Se estima que la incidencia puede ser de uno por millón de habitantes, la mayoría de las series se refieren a estudios post-mortem y al parecer se presenta con más frecuencia en Asia. Se trata de una enfermedad de la vida adulta, con un rango de edad entre los 34 y 90 años y un promedio de 70 años, afectando igualmente a hombres y mujeres (1,3-5). Los síntomas son muy variables y generalmente derivan de la oclusión de pequeños vasos por células tumorales en diferentes órganos. En las series revisadas es común el compromiso inicial del sistema nervioso central y la piel, para a medida que avanza la enfermedad comprometer órganos hematolinfoides, hígado, bazo, próstata, siendo raro el compromiso de médula ósea (1,2,5) y del pulmón, que cuando sucede, cursa como una enfermedad pulmonar intersticial o con síntomas respiratorios inespecíficos, estando reportado un caso pulmonar sin sintomatología respiratoria (6).
El compromiso del sistema nervioso central, que ocurre en cerca del 63% de los pacientes, se expresa con diversos e inespecíficos signos y síntomas, pudiéndose categorizar en cuatro grupos: eventos cerebrovasculares multifocales progresivos, síndromes medulares y de las raíces nerviosas, encefalopatía subaguda y neuropatías craneales o periféricas. De ahí la variedad de síntomas que incluyen convulsiones, confusión, demencia y otros síntomas neurológicos inespecíficos (7).
El compromiso cutáneo también es muy frecuente e inespecífico, pudiendo expresarse como erupciones eritematosas induradas, nódulos ulcerados o no, placas descamativas, celulitis, erupciones, paniculitis, talangiectasias etc. Cuando solo hay compromiso cutáneo el pronóstico es menos fatal, con una sobre vida de tres años en el 56% de pacientes, vs el 22% cuando se comprometen otros órganos. Importante resaltar que hay reportes de compromiso sistémico en ausencia de lesiones cutáneas, pero con biopsias dérmicas que demuestran células CD20 y CD45 positivas llenando los espacios vasculares, luego de previas muestras negativas. De ahí que Le et al propongan realizar biopsias dérmicas ciegas, a pacientes que clínicamente sean compatibles con este linfoma y en ausencia de manifestaciones cutáneas (8).
La expresión clínica de infiltración hematolinfoide se puede presentar como anemia, trombocitopenia y se ha descrito hepatoesplenomegalia cuando se infiltran estos dos órganos. La fiebre de origen desconocido se presenta en casi la mitad de los pacientes y las alteraciones de los paraclíncios son inespecíficas, con elevación de la deshidrogenasa láctica y la eritrosedimentación globular en la gran mayoría de los casos (2,5,9). El diagnóstico diferencial incluye diferentes neoplasias intravasculares, vasculitis del sistema nervioso central, endocrinopatías y el diagnóstico definitivo se establece mediante el estudio histológico de los órganos comprometidos y pruebas inmunohistoquímicas, que en un 60% a 80% de los casos se realiza cuando el paciente ha fallecido, pues aunque el compromiso es multiorgánico y las manifestaciones son sistémicas, difícilmente se detecta con estudios imaginológicos o exámenes de laboratorio (5,7,10).
Las biopsias de los diferentes tejidos revelan un infiltrado intravascular, sobre todo en vasos pequeños- de células grandes pleomórficas, con núcleos redondos y ovales, figuras de mitosis y escaso citoplasma. El inmunofenotipo de esta neoplasia se caracteriza por la expresión en membrana de marcadores CD20, CD79a, CD22, CD19, indicando el origen B de estas células (91%), con pocos reportes de fenotipo T (28 casos), histiocítico y NK (4 casos) (2,10,11). Recientemente se describió un caso de fenotipo citotóxico activado, en que por métodos moleculares se demostró la ausencia del receptor TCRβF1, de CD5 y expresión de CD56, que confirman el origen LTγδ del tumor y como en los reportes de casos LT, previos a este estudio, no se hizo el estudio inmunohistoquímico de marcadores citotóxicos ni de TCRβF1, los autores sugieren que dada la sobreposición de los fenotipos NK y LT, serían estudios moleculares los que en definitiva determinarían la caracterización (12).
En cuanto a la posible etiología infecciosa de este linfoma, hay reportes que tratan de relacionarlo con el virus de de Epstein-Barr (VEB), concretamente a los de origen LT, al igual que con el virus del Sida, teniendo en cuenta que en estos pacientes se ha visto incrementada la susceptibilidad a desarrollar linfomas no Hodgkin. Aunque escasos, hay reportes en la literatura de pacientes con Sida que han fallecido como consecuencia de un linfoma intravascular, demostrándose en el núcleo de las células malignas la presencia de VEB, que como se conoce, tiene escasa presencia en linfomas de origen LB. De 39 casos de linfomas intravasculares de origen LB, en sólo tres se ha encontrado VEB, dos de ellos pacientes inmunosuprimidos, mientras que de seis casos de origen LT, en tres se detectó el virus, así como en dos, de tres de origen NK, resaltando que de esos cinco positivos para VEB, sólo uno presentaba inmunosupresión. La posibilidad de que estos dos agentes pudiesen tener algún efecto en la etiología del linfoma radica en que ambos tienen origen y localización intravascular, pero se necesita de muchos y más profundos estudios para establecer esta posible relación etiológica (12, 13).
Generalmente las células neoplásicas no permean los vasos, habiéndose descrito de manera ocasional una migración a los tejidos vecinos sin formar verdaderas masas sólidas en el parénquima del órgano comprometido. Existen dos teorías fuertes que explican la preferencia de estas células por los endotelios vasculares. Una de ellas propone la expresión aberrante de marcadores de superficie CD11a y CD49D con células que preferiblemente se unen a CD54 y CD106 expresados en los endotelios. La otra teoría sugiere una pérdida en la expresión de específica de los marcadores de superficie CD29 o integrina-β-1 y de CD54 (ICAM-1) requerida para la adhesión de linfocitos y diapédesis, facilitando una migración transvascular de los linfocitos como se esquematiza en la figura 5 (8,14).

Diversos esquemas de tratamiento se han empleado en los pocos casos diagnosticados en vida y aunque es escasa la respuesta a la quimioterapia, se recomienda el uso de las diversas terapias empleadas para los linfomas ganglionares de células B. Últimamente se han utilizado los anticuerpos monoclonales anti-CD20, que parecen ser promisorios por su respuesta y baja toxicidad, con reporte de normalización de los paraclínicos y de biopsias de piel sin evidencia de linfoma en un caso (7) y de sobrevida de 24 meses en un paciente al que se le logró hacer el diagnóstico inmunohistoquímico en una biopsia de próstata (2) También se ha propuesto el transplante autólogo de células madre hematopoyéticas.
Finalmente y teniendo en cuenta que la heterogénea y a menudo sutil presentación clínica de este tumor dificultan su diagnóstico en vida y que el inicio temprano del tratamiento incrementa las posibilidades de respuesta, se ha propuesto que ante la presencia de pancitopenia, fiebre de origen desconocido, niveles elevados constantes de LDH y un cuadro leucoeritroblástico en sangre periférica, sin linfadenopatías, se podría sospechar de este tumor, impresión que se podría confirmar en forma definitiva con una biopsia cutánea, pues como se registra en la literatura, cerca de dos terceras partes de los pacientes lo presentan, incluso sin que se haga evidente el compromiso y porque la mayoría de los diagnósticos ante morten se han hecho con biopsias de piel (5,8,12).
Referencias
1. Zuckerman D. The intravascular lymphoma: the oncologist great imitator. Oncologist 2006; 11: 496-502. [ Links ]
2. Alfaro J, Espinosa A, Manríquez M, Moyano L, González N, Larrondo M. Intravascular lymphoma treated with anti CD20 monoclonal antibodies. Report of a case. Rev Med Ch 2004; 132(11):1403-06. [ Links ]
3. Sheinani K. Further evidence that "malignant angioendotheliomatosis" is an angiotropic large-cell lymphoma. New Engl J 1986; 314(15): 943 - 947. [ Links ]
4. Murase T, Nakamura S, Kawauchi K. An Asian variant of intravascular large B-cell lymphoma: clinical, pathological and cytogenetic approaches to diffuse large B-cell lymphoma associated with haemophagocytic syndrome. Br J Haematol 2000; 111: 826-834. [ Links ]
5. Yang T, Tian L, Li Q, Liu Y, Zhang Q, Ma Z, et al. A case of intravascular B-cell lymphoma presenting as myelopathy and diagnosed post mortem. J of the Neu Sci 2008; 272(1-2):196-98. [ Links ]
6. Kreiss Y, Schwartz E, Kaminski N, Raanani P, Schiby G, Zvas Z, et al. Unique pulmonary presentation of intravascular large cell lymphoma. Resp Med 1998; 92(8):1087-89. [ Links ]
7. Grove CS, Robbins PD, Kermode AG. Intravascular lymphoma presenting as progressive paraparesis J Clin Neurosc 2008; 15(9):1056-58. [ Links ]
8. Le EN, Gerstenblith MR, Gelber AC, Manno R, Ranasinghe P, Sweren RJ. The use of blind skin biopsy in the diagnosis of intravascular B-cell lymphoma. J Am Acad Dermatol 2008; 59(1):148-51. [ Links ]
9. Carter DK. Angiotropic large cell lymphoma (intravascular lymphomatosis) occurring after follicular small cleaved cell lymphoma. Mayo Clin Proc, September 1996; 71: 869 - 873. [ Links ]
10. Díaz Ll, Peniche M. Linfoma intravascular. Reporte de tres casos en el Hospital de Especialidades del Centro Médico Nacional La Raza. Patol Rev Latinoam 2008; 46 (2):120-22. [ Links ]
11. Jaffe ES, Harris NL, Stein H, Vardiman JW. Pathology and Genetics. Tumors of the Haematopoietic and Linfoid Tissues 2001; 4:177-178. [ Links ]
12. Gleason B, Brinster N, Granter S, Pinkus G, Lindeman N, Miller D. Intravascular cytotoxic T-cell lymphoma: A case report and review of the literature. J Am Acad Dermatol 2008; 58:290-4. [ Links ]
13. Merchant SH, Viswanatha DS, Zumwalt RE, Foucar K. Epstein-Barr virus Associated intravascular large T-Cell lymphoma presenting as acute renal failure in a patient with acquired Immune Deficiency Syndrome. Human Pathology 2003; 34(9):950-54. [ Links ]
14. Ponzoni M, Arrigoni G, Gould VE. Lack of CD 29 (beta1 integrin) and CD 54 (ICAM-1) adhesion molecules in intravascular lymphomatosis. Hum Pathol 2000; 31:220-226. [ Links ]

