










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Med
Print version ISSN 0121-5256On-line version ISSN 1909-7700
rev.fac.med vol.19 no.1 Bogotá Jan./June 2011
ARTÍCULO DE REVISIÓN
DISTROFIA MUSCULAR DE DUCHENNE. PERSPECTIVAS DESDE LA REHABILITACIÓN
DUCHENNE MUSCULAR DYSTROPHY. PERSPECTIVES FROM THE REHABILITATION
DISTROFIA MUSCULAR DE DUCHENNE. PERSPECTIVAS A PARTIR DA REABILITAÇÃO
DIEGO M. CHAUSTRE R. MD.a* Y WILLINGTON CHONA S. MD.a
a Residente programa de medicina física y rehabilitación, facultad de medicina, Universidad militar nueva granada, Bogotá, Colombia.
* Correspondencia: Diego Chaustre Diegomchaustrer68@yahoo.com.co. Dirección postal: Tr. 3 No 49-00, Servicio de Medicina Física y Rehabilitación, Hospital Militar Central, Bogotá, Colombia.
Recibido: Octubre 29 de 2010. Aceptado: Junio 1 de 2011.
Resumen
La Distrofia Muscular de Duchenne (DMD) es una enfermedad hereditaria de tipo neuromuscular progresiva, que se presenta durante la infancia; afecta aproximadamente 1 de cada 3500 a 6000 niños varones nacidos vivos. El gen DMD que codifica para la distrofina, una proteína estructural del músculo, se localiza en el brazo corto del cromosoma X en la región p21, puede sufrir diferentes mutaciones ocasionando la pérdida parcial o total de la proteína lo que origina la DMD y otras enfermedades relacionadas como la Distrofia Muscular de Becker, que es una distrofia de forma más leve. Esta es una enfermedad discapacitante que se caracteriza por un deterioro progresivo de los músculos, y en las etapas avanzadas de la enfermedad se presentan generalmente complicaciones cardiorespiratorias que son las causas principales que ocasionan la muerte al paciente. Desde el punto de vista de la rehabilitación, cuyo objetivo es retrasar la progresión de la enfermedad, se han venido desarrollado una serie de estrategias de tratamiento y manejo que involucran grupos multidisciplinarios; éstas van desde trabajo en gimnasio con terapeutas para estiramientos hasta prescripción de sillas de ruedas y formulación de ventilación mecánica no invasiva, que si bien se sabe no modifican las bases genéticas de la enfermedad, sí mejoran de forma importante la independencia del paciente, facilitan los cuidados por parte de la familia y los cuidadores y permiten una mejor calidad de vida.
Palabras clave: distrofia muscular de Duchenne, distrofina, rehabilitación
Abstract
Duchenne Muscular Dystrophy (DMD) is a type of severe neuromuscular disease that occurs during childhood. It affects approximately 1 in 3500 to 6000 live male births; it is associated with the recessive X chromosome, and it manifest itself in a slightest way as Becker Muscular Dystrophy, due to a mutation in the dystrophin gene located in Xp21. This is a disabling disease that inevitably occurs until a progressive deterioration of the muscles leads to the patient's death, usually from cardiorespiratory complications. From the point of view of rehabilitation, a series of management strategies have been developed in multidisciplinary groups ranging from gym work with stretch therapists to wheelchair prescriptions, and the development of noninvasive mechanical ventilation which, while they do not modify the genetic basis of the disease, do significantly improve the patient's independence as well as the care given by family and caregivers, and provide a better quality of life.
Key words: Duchenne muscular dystrophy, dystrophin, rehabilitation
Resumo
La Distrofia Muscular de Duchenne (DMD) es una enfermedad hereditaria de tipo neuromuscular progresiva, que se presenta durante la infancia; afecta aproximadamente 1 de cada 3500 a 6000 niños varones nacidos vivos. El gen DMD que codifica para la distrofina, una proteína estructural del músculo, se localiza en el brazo corto del cromosoma X en la región p21, puede sufrir diferentes mutaciones ocasionando la pérdida parcial o total de la proteína lo que origina la DMD y otras enfermedades relacionadas como la Distrofia Muscular de Becker, que es una distrofia de forma más leve. Esta es una enfermedad discapacitante que se caracteriza por un deterioro progresivo de los músculos, y en las etapas avanzadas de la enfermedad se presentan generalmente complicaciones cardiorespiratorias que son las causas principales que ocasionan la muerte al paciente. Desde el punto de vista de la rehabilitación, cuyo objetivo es retrasar la progresión de la enfermedad, se han venido desarrollado una serie de estrategias de tratamiento y manejo que involucran grupos multidisciplinarios; éstas van desde trabajo en gimnasio con terapeutas para estiramientos hasta prescripción de sillas de ruedas y formulación de ventilación mecánica no invasiva, que si bien se sabe no modifican las bases genéticas de la enfermedad, sí mejoran de forma importante la independencia del paciente, facilitan los cuidados por parte de la familia y los cuidadores y permiten una mejor calidad de vida.
Palavras-chave: distrofia muscular de Duchenne, distrofina, reabilitação
Introducción
Las distrofias musculares son un grupo de enfermedades hereditarias que se caracterizan por producir debilidad, atrofia muscular y alteración de la marcha a medida que progresa la enfermedad debido a alteraciones estructurales de las proteínas musculares que conllevan a muerte celular (1,2). Son trastornos poco frecuentes, generalmente se presentan al inicio de la infancia, siendo la forma más usual la DMD que a nivel mundial presenta una incidencia aproximada de 1 de cada 3500 a 6000 niños varones nacidos vivos (3). La DMD es una enfermedad ligada al cromosoma X de carácter recesivo; las alteraciones o mutaciones en el gen que codifica la proteína distrofina, la cual se expresa en la cara citoplasmática del sarcolema, es vital para el buen funcionamiento de las fibras musculares. La mayoría de casos de DMD están asociados a madre portadora asintomática y 1/3 de los casos se deben a mutaciones de novo (4).
Los individuos afectados pueden tener retrasos leves en el desarrollo motor y la mayoría no son capaces de correr y saltar en forma adecuada debido a la debilidad muscular proximal, lo que ocasiona la clásica maniobra de Gowers (síndrome de Gowers). La mayoría de los pacientes son diagnosticados hacia los cinco años de edad, cuando su capacidad física diverge notablemente de la de sus pares; la fuerza muscular se deteriora progresivamente y los niños requieren el uso de ayudas técnicas para la marcha y para el posicionamiento; posteriormente, en la mayoría de los casos antes de la adolescencia, requieren una silla de ruedas. Con el paso del tiempo surgen trastornos respiratorios y complicaciones cardíacas y ortopédicas, las cuales sin intervención genera una disminución considerable en la calidad de vida del paciente y de sus cuidadores. La edad media de fallecimiento de los pacientes es 18 a 19 años (5).
Hasta hace unos años, la DMD era una entidad cuyas expectativas de tratamiento eran casi nulas y aunque hasta el momento no hay una cura definitiva, existen diversas intervenciones que pueden modificar la historia natural de la enfermedad: por lo tanto actualmente, gran parte de los pacientes llegan a la edad adulta. El objetivo de este artículo es describir los principales aportes médicos para los pacientes que sufren de DMD desde el punto de vista de la medicina física y rehabilitación.
Historia
Las descripciones clínicas de la distrofia muscular de Duchenne (DMD) se han producido desde mediados de 1800. El cuadro clínico es el de una enfermedad con pérdida muscular lentamente progresiva caracterizada por síntomas que se desarrollan antes de los cinco años de edad. Inicialmente la DMD afecta los músculos de la cintura pélvica y cintura escapular, así como los músculos del cuello y los músculos abdominales, la debilidad muscular es progresiva e implacable y empeora en la infancia tardía (6).
Aunque esta enfermedad recibe su nombre del doctor Duchenne de Boulogne, importante médico y fotógrafo francés, fue Edward Meryon quien hizo las primeras descripciones de la DMD en 1852, al observar las biopsias musculares realizadas a cuatro hermanos que presentaban distrofia muscular, y reportar la presencia de alteraciones patológicas tales como disrupción del sarcolema. Para caracterizar el curso clínico realizó un informe detallado de la vida de un paciente, quien en su infancia presentaba un retraso a nivel motor y nunca pudo saltar; a la edad de ocho años tenía dificultades para subir escaleras, a los once años no podía adoptar el bípedo y a los catorce años sus extremidades superiores se habían debilitado completamente muriendo a la edad de 16 años (7).
En 1861 Duchenne describe un grupo de pacientes con paraplejía hipertrófica de gastrocnemios e inicialmente pensó que la causa era de origen cerebral; pero hacia 1868 con estudios repetidos de múltiples biopsias musculares, considera que la enfermedad es de origen muscular (8). En 1886 Gowers describe el signo clásico que lleva su nombre después observar varios niños con DMD. A partir de 1930 diferentes grupos de investigadores reportan el aumento de los niveles de enzimas musculares incluida la creatina kinasa, tanto en pacientes con diagnóstico de la enfermedad como en mujeres portadoras. Hacia 1986, Kunkel aísla el gen DMD que codifica para la proteína relacionada con la DMD y finalmente Hoffman en 1987, identifica la proteína producto de este gen que se llamaría posteriormente distrofina, molécula en la cual en la actualidad se centran las investigaciones y se espera que los resultados de esos estudios permitan diseñar estrategias para el manejo de la enfermedad (9).
Bases moleculares
El gen DMD que codifica para la distrofina se localiza en el brazo corto del cromosoma X en la región p21.2, está formado por 79 exones y tiene una extensión de 2,3 Mb. Diferentes mutaciones (deleciones, mutaciones puntuales, duplicaciones e inserciones) que ocurren en este gen son la causa de la ausencia parcial o total de la distrofina y por lo consiguiente de la DMD, así como de otras enfermedades que en conjunto se denominan distrofinopatías, como la distrofia muscular de Becker (DMB) y la cardiomiopatía dilatada 3B (10).
Para entender la patogénesis de la distrofia muscular de Duchenne (DMD) a nivel molecular y celular (9), hay que tomar en cuenta dos características importantes obtenidas a partir de las observaciones clínicas y patológicas; la primera es que, la DMD es una enfermedad crónica y debilitante, resultado de un proceso de degeneración progresiva y necrosis del músculo esquelético y cardiaco y la segunda, que la contracción muscular está asociada con la patogénesis de la DMD.
La distrofina es una proteína estructural que ayuda al anclaje de un complejo multiprotéico en la membrana celular denominado DGC (dystrophinglycoprotein complex), se encuentra principalmente en el músculo esquelético y tiene como función unir el citoesqueleto con la matriz extracelular para mantener la integridad de la membrana muscular. Su papel principal es dar soporte y protección a las células musculares durante la contracción (11); cuando la distrofina está presente, la distribución de las fuerzas mecánicas se lleva a cabo de manera coordinada en las células musculares, lo que se conoce como sistema de fijación transverso entre miofibrillas y sarcolema.
Cuando hay una deficiencia de distrofina la célula se vuelve susceptible al estrés mecánico y finalmente, la infiltración anormal de calcio (producto de la fragilidad del sarcolema) junto con la interacción de otras proteínas (calpaínas proteasas dependientes de calcio), llevan a la destrucción de la fibra muscular (12). Los músculos proximales contienen fibras grandes que soportan más peso y se ven afectadas antes que las fibras de los músculos distales. En las biopsias de pacientes con DMD es posible observar conjuntos de fibras musculares necróticas, infiltración de macrófagos y linfocitos, y fibras inmaduras con núcleos centrados, producto de ciclos de degeneración/regeneración que originan el fenotipo de la DMD (12).
Se han identificado hasta el momento varias isoformas de la distrofina: la muscular, la cortical, la cerebelar, la retiniana, la fetal (cerebro y riñón), la de la célula de Schwann y la glial (13). Aunque los síntomas de la DMD se relacionan principalmente con las isoformas musculares, se ha planteado que la ausencia de distrofina en el cerebro es la causa del déficit cognitivo observado en pacientes con DMD. También se ha demostrado que en los pacientes en quienes la mutación afecta la isoforma retiniana Dp260, presentan mayor debilidad para la visión rojo-verde. Los fibroblastos y las plaquetas expresan las isoformas Dp116 y Dp71 respectivamente; el mayor sangrado observado durante cirugías mayores se ha vinculado con las mutaciones que afectan dichas isoformas (14).
Características clínicas
Basados en los síntomas y a la progresión de la enfermedad se han descrito cinco fases:
- Fase presintomática (0 a 2 años): inicialmente el paciente no presenta características clínicas llamativas aunque puede evidenciarse un leve retraso en el desarrollo motor, sin presentarse alteraciones en la marcha. Si por los antecedentes familiares se sospecha la presencia de la enfermedad, se debe confirmar el diagnóstico con la valoración de los niveles de creatina kinasa (CK) los cuales son altos en esta entidad (15).
- Fase ambulatoria temprana (3 a 4 años): en la fase ambulatoria, los síntomas generalmente se manifiestan entre los dos y los cuatro años de edad. Los padres refieren debilidad cervical para movimientos de flexión de tronco y para mantener el equilibrio en sedente. Esta debilidad se acompaña de un retraso en el desarrollo motor. Los pacientes tienen dificultad para mantenerse a la par con sus compañeros, tanto física como cognitivamente; además es frecuente un déficit selectivo en habilidades verbales (9,16). Hacia los cinco años de edad es posible ver el característico signo descrito por Gowers para adoptar bípedo (el niño empieza a levantarse del suelo con las cuatro extremidades, se prepara para levantarse moviendo sus manos en el suelo cerca de sus pies y finalmente pone sus manos en los muslos y se impulsa para alcanzar posición bípeda) y la marcha de Trendelemburg o marcha de pato debida a debilidad de los glúteos. Además de esto se pueden observar retracciones del tendón de Aquiles y del tendón del bíceps, lo que genera mayor dificultad para caminar (1,3,17).
- Fase ambulatoria tardía (5 a 8 años): hacia los nueve años se pierde la habilidad para levantarse del piso o de una silla y subir escaleras, pero persiste la capacidad para deambular con ayudas técnicas. Las complicaciones respiratorias, gastrointestinales y cardiovasculares son infrecuentes en esta etapa (4,9).
- Fase no ambulatoria temprana (9 a 11 años): entre diez y doce años se inicia el uso de silla de ruedas, son más evidentes las contracturas en flexión de cuello de pie y tobillo, se desarrolla escoliosis progresiva (asociada su progresión a la dependencia de silla de ruedas), las actividades físicas como adoptar bípedo con ayuda pueden retardar la progresión así como ejercicios con carga de peso en agua (3,13). Los pacientes que pueden adoptar bípedo con apoyo generalmente pueden desarrollar marcha durante acuaterapia.
- Fase no ambulatoria tardía (mayores a 12 años): cambio de fase difícil de describir; son pacientes con función de extremidades superiores y mantenimiento postural cada vez más limitado, generalmente se consideran pacientes dependientes de silla de ruedas para traslados. A largo plazo presentan complicaciones serias, las más frecuentes son las que involucran el sistema respiratorio, cardiovascular y gastrointestinal que ponen en riesgo sus vidas (6,18).
Insuficiencia respiratoria: la historia natural muestra que en pacientes entre once y 20 años y que no reciben manejo con corticoesteroides, la capacidad vital forzada disminuye a 60 - 70% de la normal (19); además se presenta una disminución en la capacidad para toser y aumento de frecuencia de neumonías. La disminución de la efectividad de ventilación durante el sueño conlleva a falla respiratoria. Cabe anotar que cuando la capacidad vital forzada es menor de 1,0 L, o la PaCO2 mayor de 45 mmHg, la supervivencia estimada es de tres a cinco años (20, 21).
Cardiomiopatía: las anormalidades electrocardiográficas pueden presentarse desde la etapa ambulatoria (aumento de R-S en V1, ondas Q profundas en V5-V6); en etapas tardías se observan arritmias siendo mas frecuentes las auriculares que las ventriculares y es común encontrar taquicardia en reposo (secundaria a disfunción autonómica) (6). El ecocardiograma revela alteración primaria en función ventricular izquierda y la patología se relaciona con estos hallazgos (necrosis más importante en región posterobasal de ventrículo izquierdo).
Disfunción gastrointestinal: se observa compromiso de la musculatura voluntaria y lisa, debilidad en músculos de masticación - deglución que lleva a carraspeo y tos frecuente principalmente en el momento de alimentación, retardo en el vaciamiento gástrico y dilatación gástrica aguda que aumenta el riesgo de insuficiencia respiratoria (22). Asociado a la poca ingesta de líquidos se presenta disfunción intestinal crónica con constipación, distensión, hipomotilidad e impactación (23). Actualmente se sospecha una posible predisposición a trombosis venosas pélvicas o de miembros inferiores secundaria a distensión abdominal crónica.
Diagnóstico
La sospecha diagnóstica de la DMD debe ser considerada independientemente de la historia familiar y generalmente es accionada por diferentes hallazgos, de los cuales se deben resaltar tres. El primero y más común es la observación de función muscular anormal en niños de sexo masculino; el segundo es la detección de altos valores de la CK sérica y por último el hallazgo generalmente incidental, es un aumento de los valores de transaminasas (AST y ALT), enzimas producidas por las células hepáticas y musculares (Figura 1) (3). El diagnóstico se establece basados tantos en las características clínicas como en las paraclínicas.
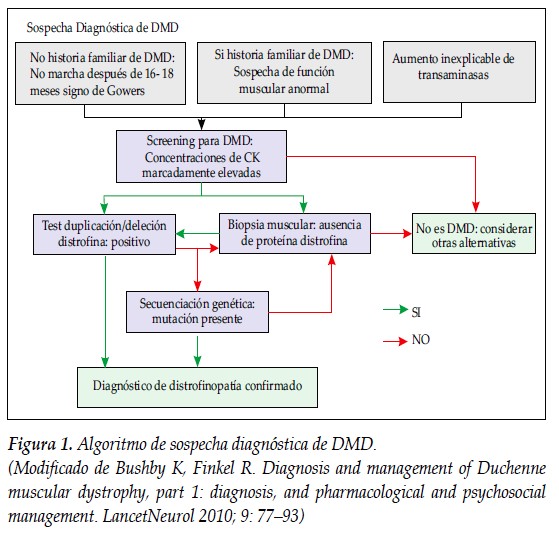
Historia clínica y examen físico
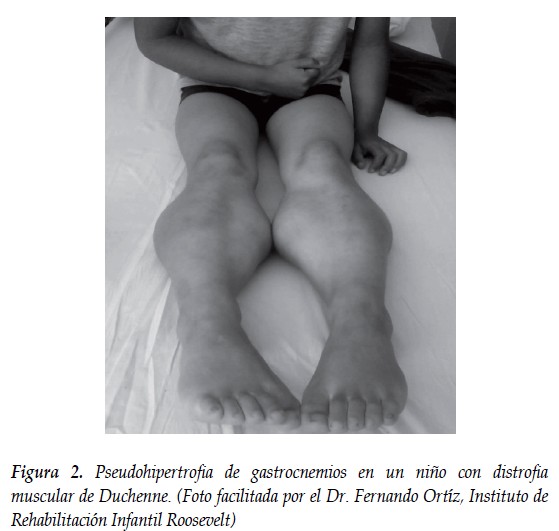
Historia familiar (algunos de los casos), función muscular anormal, retraso del desarrollo motor, caídas frecuentes, dificultad para correr y subir escaleras. Signo de Gowers, marcha de pato, pseudohipertrofia de músculos gastrocnemios (1,4) (Figura 2).
Pruebas de laboratorio: son de ayuda como diagnóstico diferencial. Desde la fase ambulatoria la CK se encuentra aumentada por lo menos diez veces del valor normal (9); también están aumentadas la AST y la ALT (17).
- Electrodiagnóstico: aunque los consensos de expertos no incluyen dentro de sus algoritmos diagnósticos el estudio con aguja de la fibra muscular, esto no indica que no pueda o deba ser realizado, especialmente si se desea descartar diagnósticos diferenciales (4). Las neuroconducciones motoras y sensitivas se hallan normales, lo hallazgos electromiográficos son: reducción de fibras musculares efectivas en una unidad motora, PAUM de corta duración y baja amplitud y contracción poco sincronizada (9).
- Biopsia muscular: puede realizarse biopsia abierta o con aguja, dependiendo del tamaño de la muestra y la severidad de la enfermedad; es importante que la muestra se tome de un músculo proximal (24). Al microscopio se encuentran fibras necróticas, infiltración de macrófagos y linfocitos y fibras inmaduras. Adicionalmente se debe realizar la detección de la distrofina por pruebas de inmnohistoquímica e inmunofluorescencia, para averiguar si existe un déficit total o parcial de la proteína (9).
- Estudio genético: hasta el momento se han detectado y registrado más de 4500 variantes del gen DMD. De estas, el 72% corresponde a deleciones extensas, el 7% a duplicaciones y el 20% a pequeñas deleciones, inserciones o mutaciones de una sola base (25).Debido a la gran cantidad de variantes para el estudio genético se han desarrollado diferentes técnicas tales como la PCR (4).
Abordaje de rehabilitación
Estiramientos
La valoración de los pacientes con diagnóstico de DMD por el médico especialista en rehabilitación es importante porque le ayuda a decidir cual es el manejo más apropiado en cada caso; lo prioritario es prevenir complicaciones musculoesqueléticas tardías y retrasar, hasta donde sea posible, la pérdida de capacidades funcionales que le impidan al paciente desempeñar las actividades de la vida diaria y su independencia (26-32). La pérdida de la capacidad funcional es principalmente el resultado de la disminución progresiva de la fuerza muscular y la resistencia muscular durante el curso progresivo de la enfermedad (31,32).
A través de diferentes estudios clínicos se ha demostrado que en el proceso de desacondicionamiento físico en personas sanas o en aquellas que han tenido periodos cortos de inactividad, ocurre una serie de procesos fisiológicos que por lo general son mínimos pero se logran recuperar rápidamente (33-39); sin embargo, esto no se observa en pacientes con enfermedades neuromusculares en quienes se tienen desenlaces devastadores y difíciles de recuperar, incluso con periodos cortos de inactividad, dando inicio así a múltiples contracturas, retracciones, deformidades vertebrales como escoliosis y/o aumento de compromiso pulmonar dado por enfermedad pulmonar restrictiva (40,41). Por estos motivos es importante que la valoración la realice un grupo multidisciplinar en el que se incluya tanto la terapia física como la ocupacional para que le ayuden al paciente a mantener la fuerza y la resistencia muscular; uno de los objetivos principales en el manejo de los trastornos neuromusculares es mantener o mejorar las características de las fibras musculares y para lo que es indispensable que se realicen ejercicios de rutina de contracción muscular contra resistencia coordinado por personal capacitado en el tema (42,43).
Un factor importante mencionado previamente es el aumento de la prevalencia de retracciones musculares con la consiguiente disminución de los Rangos de Movimiento Articular (RMA) completos, que se producen como resultado de múltiples factores entre los que se encuentran: posición estática en flexión, desequilibrio muscular y cambios fibróticos progresivos del tejido muscular (27,44, 49). Estas retracciones musculares pueden considerarse como eventos adversos que deben ser tratados y además se recomienda indicar en lo posible, el posicionamiento normal de los niños y jóvenes, permitir el movimiento, mantener la deambulación, prevenir desarrollo de deformidades fijas y mantener la piel íntegra (31,41,50). Para obtener un estiramiento efectivo de la unidad miotendinosa se requiere conjugar estiramientos activos, activos asistidos, estiramientos pasivos, sistemas de posicionamiento y órtesis (31,35,54). En niños no ambulantes adicionalmente se recomienda la movilización pasiva o ejercicios de movilización activa asistida para mantener la simetría postural (57-62).
La debilidad muscular presente en la DMD es un factor importante que contribuye al desarrollo ulterior de deformidades posturales, asimetrías de columna, escoliosis y otras posturas anómalas, que generalmente se presentan después de los seis a siete años, siendo infrecuentes en edades previas (28,30). Respecto a la prevención de contracturas, como se había mencionado anteriormente, es mandatorio realizar series de estiramientos activos, pasivos y activos asistidos con un mínimo de cuatro a seis días a la semana. Tanto en la fase ambulatoria como no ambulatoria es necesario realizar estiramiento de los músculos de las cuatro extremidades, haciendo énfasis en músculos que involucran cadera, rodilla y cuello de pie. Durante la fase no ambulatoria se debe realizar estiramiento regular de músculos de miembros superiores, haciendo énfasis en músculos relacionados con dedos, carpo, codo y hombro (29,30), la realización de estiramientos en otras articulaciones de la economía depende del examen clínico de cada paciente.
Las formas sencillas para evitar las contracturas son evitar la inmovilidad, insistir en el mantenimiento de los arcos de movilidad completos y realizar ejercicios de estiramiento (se aconsejan el yoga o taichi), acompañados de programa de terapia física y ocupacional de mínimo diez minutos y un adecuado posicionamiento para mantener los arcos de movilidad (76-78). McDonald encontró que la frecuencia de contracturas es rara antes de los nueve años, pero su frecuencia y gravedad aumenta con la edad (6). No debe olvidarse el uso de calor profundo para mejorar la calidad de los estiramientos (distensibilidad del colágeno).
Programa de ejercicio
Como se sabe que los pacientes con DMD presentan debilidad muscular progresiva secundaria a un proceso de pérdida de masa muscular por necrosis secundaria a fatiga, surge la duda acerca de si estos pacientes deben o no realizar algún tipo de ejercicio y si es así, ¿qué características debe tener el ejercicio? o ¿cuáles modalidades de los mismos se deben usar en pacientes con enfermedad de Duchenne? Se han hecho varios estudios relacionados con estas interrogantes, que han demostrado que se requiere como mínimo una intensidad del 20% de la contracción voluntaria máxima con el fin de evitar atrofia muscular por desuso, mientras que el ejercicio de fuerza y resistencia debe ser regular y con una intensidad progresiva y controlada (28,38). Jones y Rutherford (61) demostraron que incluso un programa limitado (10 repeticiones de 60% - 90% de la carga máxima/día) podría tener efectos significativos en la fuerza de los pacientes con DMD (0,5 - 1,0% aumento día durante periodo de entrenamiento de varias semanas) y finalmente después de un programa de doce semanas, la fuerza se incrementó en un 25% por unidad de superficie. Los aumentos más dramáticos de fuerza se observan en las primeras semanas, seguido por un aumento continuo pero de ritmo más lento. Estos beneficios iniciales producen Como se sabe que los pacientes con DMD presentan debilidad muscular progresiva secundaria a un proceso de pérdida de masa muscular por necrosis secundaria a fatiga, surge la duda acerca de si estos pacientes deben o no realizar algún tipo de ejercicio y si es así, ¿qué características debe tener el ejercicio? o ¿cuáles modalidades de los mismos se deben usar en pacientes con enfermedad de Duchenne? Se han hecho varios estudios relacionados con estas interrogantes, que han demostrado que se requiere como mínimo una intensidad del 20% de la contracción voluntaria máxima con el fin de evitar atrofia muscular por desuso, mientras que el ejercicio de fuerza y resistencia debe ser regular y con una intensidad progresiva y controlada (28,38). Jones y Rutherford (61) demostraron que incluso un programa limitado (10 repeticiones de 60% - 90% de la carga máxima/día) podría tener efectos significativos en la fuerza de los pacientes con DMD (0,5 - 1,0% aumento día durante periodo de entrenamiento de varias semanas) y finalmente después de un programa de doce semanas, la fuerza se incrementó en un 25% por unidad de superficie. Los aumentos más dramáticos de fuerza se observan en las primeras semanas, seguido por un aumento continuo pero de ritmo más lento. Estos beneficios iniciales producen Distrofia muscular de duchenne. Perspectivas desde la rehabilitación Volumen 19 - No. 1 - Enero - Junio de 2011 51 contracciones musculares más eficientes (34,62) mientras ganancias posteriores son secundarias a hipertrofia muscular y cambios bioquímicos (11, 35, 41).
Los niños con DMD tienen baja capacidad cardiovascular y baja utilización periférica de oxígeno, con una mayor frecuencia cardiaca en reposo en comparación con niños sanos; existen pruebas que demuestran que los niños con DMD tienen insuficiencia cronotrópica, es decir tienen disminuida la capacidad para aumentar su ritmo cardiaco en respuesta al ejercicio (42). En reposo presentan una frecuencia cardiaca 110 ± 12 latidos / minuto comparados con controles sanos (94 ± 7 latidos minuto) (86). Los estudios realizados por Bennet y Knowlton (43) muestran los riesgos que se corren al someter los músculos a exceso de trabajo (disminución de circulación, grandes esfuerzos iniciales, exigencia de fuerza máxima, gran motivación con lo que se niega el cansancio). Se debe tener en cuenta en el personal que valora y ofrece tratamientos a los pacientes con este tipo de enfermedad ya que se puede presentar fenómenos secundarios de rabdomiolisis principalmente si se realizan ejercicios de excéntricos o de estiramiento (44- 55) puesto que estos conllevan a mayor estrés mecánico de la fibra muscular que las contracciones concéntricas o de acortamiento (56, 57).
Actualmente existe un número no despreciable de estudios prospectivos que muestran la mejoría o mantenimiento de la fuerza muscular y de la resistencia en diversas enfermedades neuromusculares asociada al ejercicio; la ganancia se percibe durante la realización del ejercicio y unas semanas después de la suspensión del mismo (16, 58,61-76). Una de las manifestación de la inmovilidad son las contracturas; por eso una forma de evitarlas es insistir en el mantenimiento de los arcos de movilidad (se aconseja Yoga y Taichi) realizando programas de mínimo diez minutos y hasta seis horas de posicionamiento por día para mantener los rangos de movilidad 77-78. Otros autores recomiendan el uso de yesos pero tiene el inconveniente que debe ser realizado por personas con experiencia (79). La disminución de la capacidad aeróbica de esta enfermedad se puede atribuir a dos razones, la primera relacionada directamente con la enfermedad (involucra además la escoliosis y la enfermedad pulmonar restrictiva) y la segunda, relacionada con el nivel de actividad (42, 80-84).
Una de las formas de mejorar esta capacidad aeróbica es usar las indicaciones del Colegio Americano de Medicina del Deporte, las cuales recomiendan 15 a 60 minutos de ejercicio aeróbico, tres a cinco veces por semana, y mantener la frecuencia cardiaca entre 55% a 90% de la frecuencia cardiaca máxima (220 - edad); esta recomendación es válida para el público en general (85). Wright demostró que realizar ejercicio aeróbico de intensidad moderada mejora la capacidad aeróbica de pacientes con DMD, e incluso en pacientes con otras enfermedades neuromusculares (81,83, 84).
En conclusión se recomienda que el ejercicio en pacientes con DMD en fase ambulatoria, debe ser aeróbico sin superar el 20% de la contracción voluntaria máxima (fortalecimiento submáximo) y se deben evitar los ejercicios excéntricos, recordando que la inestabilidad inherente de la membrana del sarcolema con deficiencia de distrofina, predispone a lesiones debido a las cargas mecánicas. Los ejercicios excéntricos o contracciones de alargamiento producen más estrés mecánico en la fibra muscular que los ejercicios de contracciones concéntricas y los músculos que están sometidos a permanentemente a contracciones de alargamiento muestran la debilidad más temprana en el curso de la DMD, como los músculos extensores de cadera, extensores de rodilla y dorsiflexores del cuello de pie. En niños que no realizan deambulación se deben realizar ejercicios de movilización activa asistida con el fin de disminuir retracciones y contracturas musculares. Adicionalmente se ha encontrado que el uso de corticoides post ejercicio contribuye en la prevención de deterioro de la fibra muscular (88). Finalmente, se recomienda la incorporación de actividades recreativas y de terapia acuática, especialmente en niños preadolescentes pero de manera prudente.
Uso de silla de ruedas
La silla de ruedas es uno de las ayudas técnicas esenciales e inevitables para el paciente con DMD. La formulación oportuna del equipo adecuado puede mejorar la función y la calidad de vida de estos pacientes y sus cuidadores. Generalmente la capacidad de marcha se pierde alrededor de los 9 a 10 años de edad (1,2). La decisión de cuando usar la primera silla de ruedas así como cuando hacer la transición de una silla manual a una eléctrica es un tema de debate en la actualidad. Sin embargo, independientemente del tipo de silla de ruedas que se elija inicialmente, la mayoría de niños con DMD finalmente requieren una silla de ruedas eléctrica (116). En términos generales su uso no se requiere hasta que aparecen las caídas, dificultad extrema para levantarse desde una posición sedente o la imposibilidad de hacer ajustes posturales (117).
Durante la fase ambulatoria temprana, una silla de ruedas manual liviana se considera conveniente para permitir traslados cuando las demandas de movilidad a larga distancia son superiores a la resistencia del paciente. En la fase ambulatoria tardía es necesario el uso de una manual ultraliviana, con asiento y espalda sólido con soportes laterales para mantener el alineamiento espinal, evitar la progresión de escoliosis y adicionalmente, apoyapies en neutro para así evitar deformidades en cuello de pie. Finalmente, en la etapa no ambulatoria temprana, una silla de ruedas manual con asiento y espaldar personalizados y sistema de basculación es una condición previa a la formulación de silla de ruedas eléctrica (118-122). Si el paciente requiere una silla de ruedas eléctrica antes de indicarla, se deben tomar en consideración varios aspectos: el económico, el familiar, el del paciente (compromiso cognitivo del paciente) y el de la movilización (falta de transporte acondicionado para silla de ruedas, características del terreno, barreras arquitectónicas, etc)
Para los pacientes con DMD que físicamente no requieren una silla de ruedas eléctrica completa, una opción son las sillas con potencia en las llantas o asistidas. En estas los motores se construyen en el centro de las ruedas para ofrecer una reducción significativa del esfuerzo necesario para impulsarlas y para usar en terreno desigual, subir rampas o bajarlas, una ligera presión en las llantas que ayuda con la propulsión en cualquier dirección; en estas sillas también se recomienda asientos y componentes personalizados, incluyendo reposacabezas, asientos y espaldar sólido ajustable en altura, con soportes laterales de tronco, apoyapies y apoyabrazos a la altura del paciente. Finalmente, cuando el compromiso de las cuatro extremidades es muy severo, se pueden hacer nuevas adaptaciones a la silla de ruedas eléctrica como control con la lengua, sistemas infrarrojos o selector de mirada, todo esto para proveer la mayor independencia del paciente.
Ortesis
La prevención de contracturas y retracciones musculares son una indicación importante para el uso de ortesis, así como el adecuado posicionamiento articular y la facilitación de adopción de postura bípeda. Las Ortesis Tobillo Pie (OTP) rígidas usadas durante la noche pueden ayudar a minimizar la progresión de la contractura de cuello de pie en equino y se consideran apropiadas de por vida. Cabe recordar que estas ortesis deben ser hechas de forma individual bajo la medida de las extremidades de cada paciente para permitir el máximo confort y alineamiento del cuello de pie (123). Las Ortesis Rodilla Tobillo Pie (ORTP) pueden ser de valor durante la fase ambulatoria tardía o fase no ambulatoria temprana en la prevención de contracturas y deformidades además de permitir la posición bípeda y una deambulación limitada (con bloqueo de rodilla) que en estos casos tiene únicamente objetivos terapéuticos pero generalmente no son bien toleradas durante la noche.
Por otro lado, para los pacientes no deambuladores y que requieren desplazamiento constante en silla de ruedas, el uso de OTP durante todo el día puede ser benéfico (124). El uso de Ortesis Tobillo Pie durante el día no está indicado en pacientes en fase ambulatoria, pues generalmente limitan los movimientos compensatorios que son necesarios para una ambulación eficiente, le agregan peso al cuerpo del paciente comprometiendo la marcha y haciendo mucho más difícil la adopción de postura bípeda desde el suelo.
Referencias
1. Cammarata-Scalisi F, Camacho N. Distrofia Muscular de Duchenne, presentación clínica. Rev Chil Pediatr. 2008;79(5):495-501. [ Links ]
2. Tsao C Y, Mendell J R: Coexisting muscular dystrophies and epilepsy in children. J Child Neurol. 2006;21:148-50. [ Links ]
3. Bushby K, Finkel R. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77-93. [ Links ]
4. López-Hernández L, Vázquez-Cárdenas N. Distrofia muscular de Duchenne: actualidad y perspectivas de tratamiento. Rev Neurol. 2009;49(7):369-75. [ Links ]
5. Bushby KM, Hill A, Steele J G. Failure of early diagnosis in symptomatic Duchenne muscular dystrophy. Lancet. 1999;353:55-78. [ Links ]
6. Darras B T, Menache C C, Kunkel L M. Dystrophinopathies. In: Jones HR, DeVivo DC, Darras BT, eds. Neuromuscular Disorders of Infancy, Childhood, and Adolescence-A Clinician's Approach. Amsterdam: Butterworth Heinemann. 2003:649-700. [ Links ]
7. Meryon E. On granular and fatty degeneration of the voluntary muscles. Medico-Chirurgical Transactions. 1852;35:73-84. [ Links ]
8. Duchenne GBA. Recherches sur la paralysiemusculairepseudohypertrophiqueouparalysiemyo- sclérosique. Archives Générales de Médecine 1868; 11:5-25, 179-209, 305-321, 421-443, 552-588. [ Links ]
9. Chamberlain J, Rando T. Duchenne Muscular Distrophy, Advances in Therapeutics. Taylor and Francis Group. New York, 2006. [ Links ]
10. Hoffman E P, Brown R H Jr, Kunkel L M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919- 28. [ Links ]
11. Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;7:762-73. [ Links ]
12. Deconinck N, Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007;36:1-7. [ Links ]
13. Erazo-Torricelli R. Actualización en distrofias musculares. Rev Neurol. 2004;39:860-71. [ Links ]
14. Labarque V, Freson K. Increased Gssignalling in platelets and impaired collagen activation, due to a defect in the dystrophin gene, result in increased blood loss during spinal surgery. Hum Mol Genet. 2008;17:357-66. [ Links ]
15. Ciafaloni E, Fox D J, Pandya S. Delayed diagnosis in Duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J Pediatr. 2009;155:380-5. [ Links ]
16. Darras BT, Menache CC. Neuromuscular Disorders of Infancy, Childhood, and Adolescence-A Clinician's Approach. Amsterdam: Butterworth Heinemann. 2003:649-700. [ Links ]
17. Biggar WD. Duchenne muscular dystrophy. Pediatr Rev. 2006;27:83-8. [ Links ]
18. Hoffman E P. Dystrophinopathies. In: Karpati G, Hilton-Jones D, Griggs RC, eds. Disorders of Voluntary Muscle. Cambridge: Cambridge University Press, 2001:385-482. [ Links ]
19. Brooke MH, Fenichel GM, Griggs RC, et al. Duchenne muscular dystrophy: patterns of clinical progression and effects of supportive therapy. Neurology. 1989;39(4):475-81. [ Links ]
20. Finder JCCP. Respiratory care of the patient with Duchenne muscular dystrophy: an official ATS consensus statement. Am J Respir Crit Care Med. 2004;170(4):456-65. [ Links ]
21. Phillips M F, Quinlivan R C, Edwards R H, Calverley P M. Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am J Respir Crit Care Med. 2001;164:2191-4. [ Links ]
22. Bensen E S, Jaffe K M, Tarr P I. Acute gastric dilatation in Duchenne muscular dystrophy: a case report and review of the literature. Arch Phys Med Rehabil. 1996;77(5):512-4. [ Links ]
23. Boland B J, Silbert P L, Groover R V, Wollan P C, Silverstein M D. Skeletal, cardiac, and smooth muscle failure in Duchenne muscular dystrophy. Pediatr Neurol. 1996;14(1):7-12. [ Links ]
24. Verma S, Anziska Y. Review of Duchenne Muscular Dystrophy (DMD) for the Pediatricians in the Community. Clinical Pediatrics 49(11) 1011-7. 2010. [ Links ]
25. González-Herrera L, Gamas-Trujillo P A. Identificación de deleciones en el gen de la distrofina y detección de portadoras en familias con distrofia muscular de Duchenne/Becker. Rev Neurol. 2009;48:66- 70. [ Links ]
26. Hornyak J, Pangilinan P. Rehabilitation of Children and Adults Who Have Neuromuscular Diseases. Phys Med Rehabil Clin N Am. 2007;18:883-97. [ Links ]
27. Bushby K, Finkel R. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9:177-89. [ Links ]
28. Jansen M, De Groot M. Physical training in boys with Duchenne Muscular Dystrophy: the protocol of the No Use is Disuse study. Jansen et al. BMC Pediatrics 2010; 10:55. [ Links ]
29. Hornyak JE 4th, Pangilinan PH Jr. Rehabilitation of children and adults who have neuromuscular diseases. Phys Med Rehabil Clin N Am. 2007;18:883-97. [ Links ]
30. Bushby K, Finkel R, Birnkrant D J, Case L E, Clemens P, Cripe L. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. The Lancet Neurology. 2010;9(2):177-89. [ Links ]
31. Jansen et al. Physical training in boys with duchenne muscular dystrophy: the protocol of the no use is disuse study. BMC pediatrics. 2010. Texto online: http://www.biomedcentral.com/1471-2431/10/55 [ Links ]
32. Mannlein J, Pangilinan P. Wheelchair seating for children with Duchenne muscular dystrophy , journal of pediatric rehabilitation medicine: an interdisciplinary approach. 2008;1(3):225-35. [ Links ]
33. Duchenne muscular dystrophy Advances in therapeutics; published in 2006 by Taylor and Francis Group , 270 madison avenue, new york, ny 10016 [ Links ]
34. Mcdonald C M, Carter G, Fowler W et al. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1995;74:70-92. [ Links ]
35. Bar-or O, Rowland TW: Neuromuscular and musculoskeletal diseases. In Pediatric Exercise Medicine From Physiologic Principles to Health Care Application. Edited by: Bar-or O, Rowland W. Human Kinetics; 2004:269-303. [ Links ]
36. Akeson W, Amiel D, Abel M, Garfin S, Woo S. Effects of immobilization on joints. Clinical Orthopaedics and Related Research. 1987;219:28-37. [ Links ]
37. Booth F W. Physiologic and biochemical effects of immobilization on muscle. Clinical orthopaedics and related research. 1987;219:15-9. [ Links ]
38. Colliandereb T. Effects of detraining following short term resistance training on eccentric and concentric muscle strength. Acta Physiol Scand. 1992;144:23-9. [ Links ]
39. Hainaut K, Duchateau J. Muscle fatigue, effects of training and disuse. Muscle & Nerve. 1989;12:660-9. [ Links ]
40. Kottkef J. The effects of limitation of activity upon the human body. JAMA. 1966;196:117-22. [ Links ]
41. Muller E A. Influence of training and of inactivity on muscle strength. Arch Phys Med Rehabil. 1970;51:449-62. [ Links ]
42. Robinson G A, Stuart D G. Immobilization-induced changes in motor unit force and fatigability in the cat. Muscle & Nerve. 1991;14:563-73. [ Links ]
43. Bach J, Mckeon J. Orthopedic surgery and rehabilitation for the prolongation of bracefree ambulation of patients with duchenne muscular dystrophy. American Journal of Physical Medicine and Rehabilitation. 1991;70:323-31. [ Links ]
44. Fowler W M. Rehabilitation management of muscular dystrophy and related disorders: i. The role of exercise. Arch Phys Med Rehabil. 1982;63:319-21. [ Links ]
45. Gardner-medwin D. Management of muscular dystrophy. Physiotherapy. 1977;63:46-55. [ Links ]
46. Harris S E, Cherry D B. Childhood progressive muscular dystrophy and the role of physical therapy. Physical Therapy. 1974;54:4-12. [ Links ]
47. Brooke M H, Fenichel G M, Griggs R C et al. Duchenne muscular dystrophy: patterns of clinical progression and effects of supportive therapy. Neurology. 1989;39:475-81. [ Links ]
48. Johnson E W, Walter J. Zeiter lecture: pathokinesiology of duchenne muscular dystrophy: implications for management. Arch Phys Med Rehabil. 1977;58:4-7. [ Links ]
49. Sutherland D H, Olshen R, Cooper l, et al. The pathomechanics of gait in duchenne muscular dystrophy. Dev Med Child Neurol. 1981;23:3-22. [ Links ]
50. Archibald K C, Vignosp J. A study of contractures in muscular dystrophy. Arch phys med rehabil. 1959;40:150-57. [ Links ]
51. Johnson E R, Fowler W, Lieberman J. Contractures in neuromuscular disease. Arch Phys Med Rehabil. 1992;73:807-10. [ Links ]
52. Hsu J D, Furumasu J. Gait and posture changes in the duchenne muscular dystrophy child. Clin Orthop Relat Res. 1993;288:122-25. [ Links ]
53. Dubowitz V. Progressive muscular dystrophy: prevention of deformities. Clin Pediatr. 1964;12:323-8. [ Links ]
54. Dubowitz V. Prevention of deformities. Isr J Med Sci. 1977;13:183-8. [ Links ]
55. Vignosp J. Physical models of rehabilitation in neuromuscular disease. Muscle & Nerve. 1983;6:323-38. [ Links ]
56. Siegel I M, Weiss L A. Postural substitution in duchenne's muscular dystrophy. JAMA. 1982;247:584. [ Links ]
57. Owler W. Rehabilitation management of muscular dystrophy and related disorders. Comprehensive care. Arch Phys Med Rehabil. 1982;63:322-8. [ Links ]
58. Hyde S A, Fløytrup I, Glent S et al. A randomized comparative study of two methods for controlling tendoachilles contracture in duchenne muscular dystrophy. Neuro Muscul Disord. 2000;10:257-63. [ Links ]
59. Johnson E W, Kennedy J. Comprehensive management of duchenne muscular dystrophy. Arch Phys Med Rehabil. 1971;52:110-14. [ Links ]
60. Eagle M, Baudouin S V, Chandler C, Giddings D, Bullock R, Bushby K. Survival in duchenne muscular dystrophy: improvements in lifeexpectancy since 1967 and the impact of home nocturnal ventilation. Neuro Muscul Disord. 2002;12:926-9. [ Links ]
61. Jones D A, Rutheford O. Human muscle strength training: the effects of three different regimes and the nature of the resultant changes. J Physiol. 1987;391:1-11. [ Links ]
62. Edstrom L, Grimby L. Effect of exercise on the motor unit. Muscle & Nerve. 1986;9:104-26. [ Links ]
63. Alway S E, Macdougall J, Sale D et al. Functional and structural adaptations in skeletal muscle of trained athletes. J Appl Physiol. 1988;64:1114-20. [ Links ]
64. Cote C, Simoneau J, Lagasse P et al. Isokinetic strength training protocols: do they show 132induce skeletal muscle fiber hypertrophy? Arch Phys Med Rehabil. 1988;69:281-5. [ Links ]
65. Herbison G et al. Exercise of normal muscle: biochemical effects. Arch Phys Med Rehabil. 1973;54:409-15. [ Links ]
66. Luthi J, Howald H, Claassen H et al. Structural changes in skeletal muscle tissue with heavyresistance exercise. Int J Sports Med. 1986;7:123-7. [ Links ]
67. Adams M A, Chandler L. Effects of physical therapy program on vital capacity of patients with muscular dystrophy. Physical Therapy. 1974;54:494-6. [ Links ]
68. Hortobagyi T, Katch F. Role of concentric force in limiting improvement in muscular strength. J Appl Physiol. 1990;68:650-8. [ Links ]
69. Holloszy J. Muscle metabolism during exercise. Arch Phys Med Rehabil. 1982;63:231-4. [ Links ]
70. Sockolov R, Irwin B, Dressendorfer R, Fowler W. Exercise performance in 6 to 11 year old boys with duchenne muscular dystrophy. Arch Phys Med Rehabil. 1977;58:195-201. [ Links ]
71. Bennett R, Knowlton G. Overwork weakness in partially denervated skeletal. Muscle Clin Orthop. 1958;22-9. [ Links ]
72. Balnave C, Thompson M. Effect of training on eccentric exerciseinduced muscle damage. J Appl Physiol. 1993;75:1545-51. [ Links ]
73. Clarkson P, Kazuno R, Braun B. Muscle function after exerciseinduced muscle damage and rapid adaptation. Med Sci Sports Exerc. 1992;24:512-20. [ Links ]
74. Duchateau J, Hainaut K. Isometric or dynamic training: differential effects on mechanical properties of a human muscle. J Appl Physiol. 1984;56:296-301. [ Links ]
75. Hather B, Tesch P, Buchanan P et al. Influence of eccentric actions on skeletal muscle adaptations to resistance training. Acta Physiol Scand. 1991;143:177-85. [ Links ]
76. Byrd S K. Alterations in the sarcoplasmic reticulum: a possible link to exercise-induced muscle damage. Med Sci Sports Exerc. 1992;24:531-6. [ Links ]
77. Clarkson P, byrneswc, mccormick km, et al. Muscle soreness and serum creatine kinase activity following isometric, eccentric, and concentric exercise. Int J Sports Med. 1986;7:152-7. [ Links ]
78. Clarkson P, Tremblay I. Exercise-induced muscle damage, repair, and adaptation in humans. J Appl Physiol. 1988;65:1-6. [ Links ]
79. Friden j, lieberrl. Structural and mechanical basis of exercise-induced muscle injury. Med sci sports exerc 1992;24:521-30. [ Links ]
80. Leiber R, Friden J. Muscle damage is not a function of muscle force but active muscle strain. J Appl Physiol. 1993;74:520-6. [ Links ]
81. Mccully K, Shellock F, Bank W et al. The use of nuclear magnetic resonance to evaluate muscle injury. Med Sci Sports Exerc. 1992;24:537-42. [ Links ]
82. Mcneil P, Khakee R. Disruptions of muscle fiber plasma membranes: role of exerciseinduced damage. Am J Path. 1992;140:1097-109. [ Links ]
83. Sacco P, Jones D, Dickjrt et al. Contractile properties and susceptibility to exercise-induced damage of normal and mdx mouse tibialis anterior muscle. Clin Sci. 1992;82:227-36. [ Links ]
84. Carter G, Kikuchi N, Abresch R, Walsh S, Horasek S, Fowler W. Effects of exhaustive concentric and eccentric exercise on murine skeletal muscle. Arch Phys Med Rehab. 1994;75(5):555-9. [ Links ]
85. Carter G, Wineinger M, Walsh S, Horasek S, Fowler W. Effect of voluntary wheel-running exercise on muscles of the mdx mouse. Neuro muscul disord. 1995;5(4):323-31. [ Links ]
86. Aitkens S, Mccrory M, Kilmerd D et al. Moderate resistance exercise program: its effect in slowly progressive neuromuscular disease. Arch Phys Med Rehabil. 1993;74:711-5. [ Links ]
87. Bohannon R, Jones P. Results of manual resistance exercise on a manifesting carrier of duchenne muscular dystrophy: A case report. Phys Ther. 1986;66:973-5. [ Links ]
88. Delateur B, Giaconi R. Effect on maximal strength of submaximal exercise in duchenne muscular dystrophy. Am J Phys Med. 1979;58:26- 36. [ Links ]
89. Dimarco A F, Kelling J S, Dimarco M et al. The effects of inspiratory resistive training on respiratory muscle function in patients with muscular dystrophy. Muscle & Nerve. 1985;8(4):284-90. [ Links ]
90. Dupont-versteegden E, Mccarter R, Katz M. Voluntary exercise decreases progression of muscular dystrophy in diaphragm of mdx mice. J Appl Physiol. 1994;77:1736-41. [ Links ]
91. Dscalante A, Miller L, Beardmore T. Resistive exercise in the rehabilitation of polymyositis/ dermatomyositis. J Rheum. 1993;20:1340-4. [ Links ]
92. Florence J, Hagberg J M. Effect of training on the exercise responses of neuromuscular disease patients. Med Sci Sports Exerc. 1984;5:460-5. [ Links ]
93. Hicks J, Miller F, Plotz P, et al. Isometric exercise increases strength and does not produce sustained creatinine phosphokinase increases in a patient with polymyositis. J Rheu. 1993;20:1399-401. [ Links ]
94. Kawazoe Y, Kobayashi M, Tasaka T et al. Effects of therapeutic exercise on masticatory function in patients with progressive muscular dystrophy. J Neurol Neurosurg Psychiatry. 1982;45:343-7. [ Links ]
95. Kilmer D, Mccrory M, Wright N et al. The effect of high resistance exercise program in slowly progressive neuromuscular disease. Arch Phys Med Rehabil. 1994;75:560-3. [ Links ]
96. Iohi E, Lindber C, Andersen O. Physical training effects in myasthenia gravis. Arch Phys Med Rehabil. 1993;74:1178-80. [ Links ]
97. Mccartney N, Moroz D, Garner S et al. The effects of strength training in show 132 patients with selected neuromuscular disorders. Med Sci Sports Exerc. 1988;20:362-8. [ Links ]
98. Milner-brown H, Miller R. Muscle strengthening through highresistance weight training in patients with neuromuscular diseases. Arch Phys Med Rehabil. 1988;69:14-9. [ Links ]
99. Martin R, Sufit R, Ringle S et al. Respiratory improvement by muscle training in adultonset acid maltase deficiency. Muscle & Nerve. 1983;6:201-3. [ Links ]
100. Scott O, Hyde S, Goddard C et al. Effect of exercise in duchenne muscular dystrophy. Physiotherapy. 1981;67:174-6. [ Links ]
101. Spector S, gordonpl, et al. Strength gains without muscle injury after strength training with postpolio muscular atrophy. Muscle & Nerve. 1996;19:1282-90. [ Links ]
102. Spector S, Lemmer J, Koffman B et al. Safety and efficacy of strength training in show 132 patients with sporadic inclusion body myositis. Muscle & Nerve. 1997;20:1242-8. [ Links ]
103. Taylor R, Fowler W, Doerr l. Exercise effect on contractile properties of skeletal muscle in mouse muscular dystrophy. Arch Phys Med Rehabil. 1976;57:174-80. [ Links ]
104. Vignos P, Watkins M. The effect of exercise in muscular dystrophy. JAMA. 1966;197:121-6. [ Links ]
105. Webster C, Silberstein L, Hays A, Blau H. Fast muscle fibers are preferentially affected in duchenne muscular dystrophy. Cell. 1988;52(4):503-13. [ Links ]
106. Tardieu C, Lespargot A, Tabary C et al. For how long must the soleus muscle be stretched each day to prevent contracture? Developmental medicine and child neurology. 1988;30:3-10. [ Links ]
107. Cusick B. Splints and casts: managing foot deformity in children with neuromotor disorders. Physic Ther. 1988;68:1903-12. [ Links ]
108. Bar-or O. Role of exercise in the assessment and management of neuromuscular disease in Children. Med Sci Sports Exerc. 1995;28:421-7. [ Links ]
109. Edwardsr H. Studies of muscular performance in normal and dystrophic subjects. Brit Med Bull. 1980;36:159-64. [ Links ]
110. Haller R, Lewis S. Pathophysiology of exercise performance in muscle disease. Med Sci Sports Exerc. 1984;16:456-9. [ Links ]
111. Lewis S. Exercise and human neuromuscular diseases: a symposium overview. Med Sci Sports Exerc. 1984;16:449-50. [ Links ]
112. Wright N, Kilmer D, Mccrory M et al. Aerobic walking in slowly progressive neuromuscular Disease: effect of a 12-week program. Arch Phys Med Rehabil. 1996;77:64-9. [ Links ]
113. American college of sports medicine. In: guidelines for exercise testing and prescription. 4th ed. Philadelphia: lea & febiger; 1991. Páginas 11-32. [ Links ]
114. Mcdonald C M, Widman L, Walsh D, Walsh S, Abresch R. Use of step activity monitoring for continuous physical activity assessment in boys with duchenne muscular dystrophy. Arch Phys Med Rehabil. 2005;86:802-8. [ Links ]
115. Carter G T, Abresch R, Fowler W. Adaptations to exercise training and contraction-induced muscle injury in animal models of neuromuscular disease. Am J Phys Med Rehabil. 2002;81:151-161. [ Links ]
116. Lim J, Kim D, Bang M. Effects of exercise and steroid on skeletal muscle apoptosis in the mdx mouse. Muscle & Nerve. 2004;30:456-62. [ Links ]
117. Bush B. The multidisciplinary management of duchennemuscular dystrophy, Curr Pediat. 2005;15(4):292-300. [ Links ]
118. Pellegrini N, Guillon B, Prigent H et al., optimization ofpower wheelchair control for patients with severe duchennemuscular dystrophy, Neuro Muscul Disord. 2004;14(15):297-300. [ Links ]
119. Clark J, Michael S, Morrow M. Wheel chair postural support For young people with progressive neuromuscular disorders. Int J Therapy Rehabil. 2004;11(8)365-373. [ Links ]
120. Engstrom E D. Ergonomics, wheelchairs and positioning, Sweden: brommatryck, ab; 1993 [ Links ]
121. Bakker J, de Groot I J, Beelen A, Iankhorst G. Predictive factors of cessation of ambulation in patients with duchenne muscular dystrophy, Am J Phys Med Rehabil. 2002;81(12):906-12. [ Links ]
122. Guerette P, Tefft D, Furumasu J. Pediatric powered wheelchairs: results of a national survey of providers. Assist Technol. 2005;17(2):144-58. [ Links ]
123. Mcdonald C. Limb contractures in progressive neuromuscular disease and the role of stretching, orthotics, and surgery. Phys Med Rehabil Clin N Am. 1998;9:187-211. [ Links ]
124. Siegel I. Plastic-molded knee-ankle-foot orthoses in the treatment of duchenne muscular dystrophy. Arch Phys Med Rehabil. 1975;56:322. [ Links ]
125. Bakker J, de Grooti J, de Jong B et al. Prescription pattern for orthoses in the netherlands: use and experience in the ambulatory phase of duchenne muscular dystrophy. Disabil Rehabil. 1997;19:318-25. [ Links ]

