










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Med
Print version ISSN 0121-5256On-line version ISSN 1909-7700
rev.fac.med vol.19 no.2 Bogotá July/Dec. 2011
ARTÍCULO DE REVISIÓN
CARCINOMA CORTICOADRENAL. REVISIÓN DE TEMA
ADRENOCORTICAL CARCINOMA - SUBJECT REVIEW
ADENOCARCINOMA CORTICOADRENAL. REVISÃO DE TEMA
DIANA POLANÍAa, JORGE GARCÍA LINARESb, CARLOS GARCÍA LINARESc, PILAR CRISTINA RUIZd, OLGA MERCEDES MEJÍA RIVERAe
a Endocrinóloga. Profesora de endocrinología del HOMIC.
b Endocrinólogo. Profesor de endocrinología del HOMIC.
c Endocrinólogo. Jefe de endocrinología del HOMIC.
d Endocrinóloga. Profesora de endocrinología del HOMIC.
e Fellow de endocrinología del HOMIC.
* Correspondencia: Olgamercy2003@yahoo.es. Celular: 3006160294
Recibido: noviembre 2 de 2011
Aceptado: diciembre 28 de 2011
Introducción
Los adenomas son los tumores corticoadrenales más comunes, con una prevalencia del 4%, siendo los carcinomas corticoadrenales raros, con una incidencia de 1 caso por millón de habitantes. La mitad de estos carcinomas son funcionales con diagnósticos más tempranos por los síndromes de exceso de hormonas que producen, a diferencia del restante 50% que es silente, con hallazgo de metástasis al momento del diagnostico en más de la mitad de ellos. Pueden tener una presentación esporádica o hacer parte de síndromes de cáncer familiar con anormalidades en genes que resultan en un imbalance entre oncogenes y genes supresores de tumor; además de constituirse como potenciales marcadores diagnósticos, pronósticos y terapéuticos de esta enfermedad. Ante la sospecha de carcinoma de corteza suprarrenal, deben tenerse en cuenta criterios clínicos, bioquímicos y radiológicos para establecer adecuadamente el diagnóstico, el cual debe confirmarse por histología.
Palabras clave: carcinoma, fisiopatología, alteraciones genéticas, estadiaje, terapia, seguimiento.
Introduction
Adenomas are the most common adrenocortical tumors with a prevalence of 4%, being the adrenocortical carcinomas rare with an incidence of 1 case por million people. Half these carcinomas are functional, with earlier diagnoses due to the excessive hormonal syndromes they produce unlike the remaining 50% which are silent, with metastases being found upon the diagnosis in more than one half of them. They can have a sporadic presentation or make part of familiar cancer syndromes with genetic disturbances resulting in an imbalance between the oncogenes and the tumor suppressor genes, aside from becoming potential diagnostic, prognostic and therapeutic markers of this disease. In the presence of adrenocortical carcinoma, several clinical, biochemical and radiological criteria must be taken into account to properly establish the diagnosis which should be confirmed through histology.
Key words: carcinoma, pathophysiology, genetic disturbances, staging, therapy, follow-up.
Introdução
Os adenomas são os tumores corticoadrenais mais comuns, com uma prevalência de 4%, sendo os adenocarcinomas corticoadrenais raros, com una incidência de 1 caso por milhão de habitantes. A metade destes carcinomas são funcionais com diagnósticos mais precoces pelas síndromes de excesso de hormônios que produzem, ao contrário do 50% restantes que é silente, com descobrimento de metástase no momento do diagnostico em mais da metade deles. Podem ter uma apresentação esporádica ou fazer parte de síndromes de câncer familiar com anormalidades em genes que resultam em um desequilíbrio entre oncogenes e genes supressores de tumor; além de constituir-se como potenciais marcadores diagnósticos, prognósticos e terapêuticos desta doença. Diante da suspeita de carcinoma do córtex suprarrenal, devem ser levados em consideração critérios clínicos, bioquímicos e radiológicos para estabelecer adequadamente o diagnóstico, que deve ser confirmado por histologia.
Palavras-chave: carcinoma, fisiopatologia, alterações genéticas, estadia, terapia, acompanhamento.
Caso clínico HOMIC
Mujer de 46 años, en tratamiento para artritis reumatoidea temprana, con hallazgo incidental en ecografía de masa abdominal, solicitada para estudio de trombocitopenia. TAC abdomen: "Masa localizada en hipocondrio y flanco izquierdos, contornos bien definidos de densidad no homogénea por zona de necrosis hacia la margen inferior, de 10.4 x 10 cm con una extensión de 17 cm...". Sin síntomas constitucionales ni sugestivos de endocrinopatía. Intervenida quirúrgica/ en mayo/11, extraen masa retroperitoneal de 693 gms, cuya patología reporta carcinoma adrenal, sin invasión capsular ni vascular. Índice mitótico mayor de 5 por 50 campos de alto poder. Inmunohistoquímica: Positivo para vimentina, S100-sinaptofisina y melan A. Negativo para cromogranina, enolasa, P53, AE1/AE3. Ki67 no contributivo. En junta de tumores deciden manejo con radioterapia 30 sesiones.
Epidemiología
Se trata de una patología rara, con una incidencia de 0.7 a 2 personas por millón de habitantes (1). La incidencia más alta en el mundo se ha encontrado en niños en el sur de Brasil: 3.4 a 4.2 por millón de niños vs 0.3 por millón de niños en el resto del mundo; en más del 80% de ellos se ha evidenciado una mutación germinal especifica en el gen TP53, que se manifiesta exclusivamente en esta población.
En Colombia, no hay datos epidemiológicos específicos, pero según registros del Instituto Nacional de Cancerología, los tumores malignos de la glándula adrenal (incluidos los originados en la corteza y en la medula), representan 0.1% de los diagnósticos nuevos de neoplasia maligna (2).
La edad de presentación tiene una distribución bimodal, con picos en la primera y cuarta décadas de la vida y una edad promedio de presentación en el adulto a los 45 años. Es ligeramente más frecuente en mujeres que en hombres, con una relación de 1.5; como se ilustro en nuestro caso.
Varias series de casos describen una ocurrencia mayor de carcinoma adrenocortical en el lado izquierdo (55% vs 45%), que también sucedió en la paciente del caso (3).
El tumor puede tener una presentación esporádica o ser parte de síndromes familiares de cáncer como los síndromes de Li Fraumeni y de Beckwith Wiedemann entre otros (4).
Cerca de la mitad de estos tumores son funcionales y cursan con síndromes característicos que alertan el diagnostico; el otro 50% es silente y el diagnostico se hace generalmente en estados avanzados con metástasis a distancia en más de la mitad de pacientes y sobrevida menor a un año en la mayoría de los casos. (1, 5).
Fisiopatología y alteraciones genéticas
Una fuerte evidencia demuestra alteraciones genéticas casi constantes en estos tumores; así, muchos tumores originalmente considerados esporádicos, podrían tener también bases heredables.. La mayoría de estos genes corresponden a oncogenes o a genes supresores de tumores; su sobreexpresión y/o su subexpresión respectivamente, explican algunos de los síndromes de cáncer familiar (6).
Oncogenes
La activación constitutiva de ß-catenina, mediante mutaciones que disminuyen o evitan su fosforilación, es la alteración más frecuente en TACs benignos y malignos.
Acumulación anormal de ß-catenina, se detectó en 10 de 26 (36%) adenomas y en 11 de 13 (85%) carcinomas. (7)
El gen IGF2 está localizado en el cromosoma 11p15, región 15.5; esta región está dividida en 2 dominios funcionales: Dominio 1 contiene 2 genes IGF2 y H19. Normalmente IGF2 es expresado únicamente del alelo paterno y el H19, el cual modula la expresión de IGF2, es expresado sola/ del alelo materno. Cuando se pierde el alelo materno y hay duplicación del alelo paterno, fenómeno llamado isodisomía paterna, se presenta una sobre expresión de IGF2, el cual actúa como un factor de crecimiento autocrino a través de la unión al receptor IGF1 promoviendo proliferación de células de carcinoma adrenocortical. El dominio 2 contiene varios genes que incluyen, KCNQ1, KCNQ1OT1 y CDKN1c (p57 kip), este último gen codifica un inhibidor de kinasa dependiente de ciclina, su sobre expresión detiene el ciclo celular en la fase G1 y viceversa (9).
Análisis del locus 11p15 es útil en tumores adrenocorticales (TACs), ya que la perdida de heterocigocidad es significativamente más frecuente en carcinoma que en adenoma benigno (78.5% vs 9.5%) y está asociado con un riesgo más alto de recurrencia tumoral.
BWS es uno de los trastornos de sobre crecimiento más frecuentes; ocurre 1:14000 recién nacidos y es clásicamente caracterizado por: exoftalmos, macroglosia, gigantismo e hiperplasia con asimetría entre los lados derecho e izquierdo del cuerpo (10). La hipoglicemia ocurre en 50% de ellos y es causada por hiperplasia de células de islotes pancreáticos. Adicionalmente presentan visceromegalia de hígado, páncreas, bazo riñones y adrenales. Estos pacientes tienen una predisposición alta a desarrollar neoplasias malignas embrionarias incluyendo tumor de Wilms, rabdomiosarcoma y tumores adrenocorticales (11).
Factor esteroidogénico 1 (SF1)
SF1 juega un papel importante en el desarrollo adrenal (12). Se ha demostrado su sobreexpresión en la mayoría de casos de TACs de comienzo en la infancia y también en muchos casos de inicio en la adultez (13).
Factores de crecimiento
Varios factores de crecimiento y/o citokinas diferentes aIGF2, también regulan el crecimiento y la función normal de la glándula adrenal adulta y fetal. Se ha encontrado una mayor expresión del factor de crecimiento endotelial vascular (VEFG) en carcinomas adrenocorticales que en adenomas y en adrenales sanas (14).
Genes supresores de tumor
TP53 - Síndrome de Li Fraumeni
P53 es una fosfoproteína de 53 kDa, codificada por un gen localizado en el brazo corto del cromosoma 17.. P53 bloquea progresión del ciclo celular en las fases G1 y G2, por lo tanto previene la replicación de lesiones en el DNA potencialmente promotoras de tumor y la división de células anormales. Adicionalmente, P53 activado, gatilla apoptosis de células dañadas (15).
Mutaciones germinales en P53, están presentes en 70% de familias con síndrome de Li Fraumeni (16). Este síndrome confiere susceptibilidad a otros tumores: Cáncer de mama, sarcoma de tejidos blandos, tumores cerebrales, osteosarcoma, leucemia y cáncer corticoadrenal. Estos tumores tienen comienzo temprano y afectan casi siempre a niños y adultos jóvenes.
Estas mutaciones germinales en P53, han sido vistas en 50 a 80% de niños con cáncer adrenocortical aparentemente esporádico. Hay un grupo especial de casos de TAC infantil en el sur de Brasil, donde la incidencia de estos tumores es 15 veces mayor que en el resto del mundo. Más del 80% de estos pacientes, portan una mutación germinal P53 específica (R337H), solo reportada en esta población; sus familias no presentan el perfil clásico de síndrome Li Fraumeni (1, 17).
En adultos, se han encontrado mutaciones somáticas de P53 en 20 - 35% de casos de cáncer adrenocortical esporádico y podrían estar asociados con tumores más agresivos y avanzados.
La pérdida de heterocigocidad en 17p13, ha sido reportada más frecuentemente en pacientes con carcinoma (85%) que en adenoma (30%). Además es un marcador predictivo independiente, de recurrencia, después de la remoción quirúrgica del tumor (1, 9).
Datos de la Agencia Internacional para la Investigación del Cáncer, reportan que 80% de pacientes con cáncer de mama que portan mutaciones P53, pertenecen a una familia con síndrome de Li Fraumeni; en esos casos las mutaciones típicamente se manifestaron por P53 inactiva. En cambio, solo 15% de niños con TACs, son de familia con síndrome Li Fraumeni; muchos de estos niños portan mutaciones P53 de baja penetrancia que codifican proteína P53 parcialmente funcional (18).
Niños con TAC, independiente de su historia familiar de cáncer, deben ser considerados portadores de mutaciones germinales P53 (19).
Receptor de melanocortina 2 (MC2R)
Sus genes están localizados en el cromosoma 18p11.2. Se sugiere un papel en la diferenciación familiar. La expresión de RNAm de MC2R está regulado en alto en adenomas funcionales y regulados en baja en adenomas no funcionales y carcinomas (20).
Manifestaciones clínicas
El cuadro clínico depende de la funcionalidad y del tamaño del tumor.
En el carcinoma adrenocortical funcionante, el síndrome de Cushing es la presentación más frecuente, el cual produce un patrón clínico alterado dado su desarrollo rápido, con poca o ninguna ganancia de peso, atrofia muscular severa, HTA de difícil control y DM como las características dominantes. También puede presentarse hipokalemia severa por la activación de los receptores mineralocorticoides al ser sobrepasada la capacidad de la 11ß-deshidrogenasa-2 para inactivar el cortisol. Un porcentaje alto de mujeres afectadas desarrollan signos de virilización con o sin síndrome de Cushing concomitante (21).
Los otros síndromes de hipersecreción hormonal son menos frecuentes e incluyen exceso de producción de estrógenos y de aldosterona. En el caso de producción tumoral de estrógenos en hombres, va a producir feminización, con ginecomastia de inicio reciente, pérdida de la libido y atrofia testicular; en una serie alemana de 194 hombres con carcinoma adrenocortical, 14 (7%) presentaron feminización (22). El carcinoma productor de aldosterona es muy raro y se manifiesta con HTA e hipokalemia severa. Hipoglicemia inducida por tumor, ha sido repetidamente descrita en estos pacientes, probablemente refleje el aumento en la utilización de glucosa inducida por liberación paracrina de IGF2 (23).
La clínica de los tumores no funcionales se relaciona con el efecto de masa abdominal que producen, presentándose usualmente con náuseas, vómito, llenura o dolor dorsal. En raras ocasiones se puede manifestar como hemorragia retroperitoneal, por ruptura espontánea del tumor. Estos tumores no funcionales también pueden presentarse por la presencia de metástasis, los sitios más frecuentes son, hígado, pulmón y ganglios linfáticos (24).
Un porcentaje en ascenso de este carcinoma, se presenta como incidentaloma, dado el uso cada vez mayor de imágenes diagnosticas. En una serie del registro de cáncer adrenocortical alemán, 17.7% de 581 pacientes, se encontraron como incidentalomas (3,22).
Diagnóstico
Ante la sospecha de carcinoma de corteza suprarrenal, deben tenerse en cuenta criterios clínicos, bioquímicos y radiológicos para establecer adecuadamente el diagnóstico, el cual debe confirmarse por histología (2).
Pruebas bioquímicas
Los estudios bioquímicos incluyen un perfil completo de hormonas esteroideas; aunque el estado funcional del tumor no se correlaciona con el potencial de malignidad, estas medidas son útiles como marcadores tumorales durante el seguimiento para detección temprana de recurrencia e importantes para definir el mejor tratamiento para esta enfermedad. Además, la demostración de exceso de glucocorticoides antes de la cirugía, es esencial para prevenir crisis adrenales postoperatorias. Todos los pacientes con sospecha de carcinoma corticoadrenal, deben tener una prueba de supresión con dexametasona a dosis bajas a la medianoche, para exclusión de hipercortisolismo, aun en ausencia de clínica de síndrome de Cushing. De igual manera es importante la exclusión de manera temprana de feocromocitoma, para evitar diagnósticos erróneos y complicaciones intraoperatorias inesperadas y potencialmente fatales (1).
A continuación se presentan los exámenes hormonales pertinentes en pacientes con diagnóstico sospechoso o confirmado de carcinoma adrenocortical, según las recomendaciones de la Red Europea para el estudio de Tumores Adrenales (ENSAT). (25)
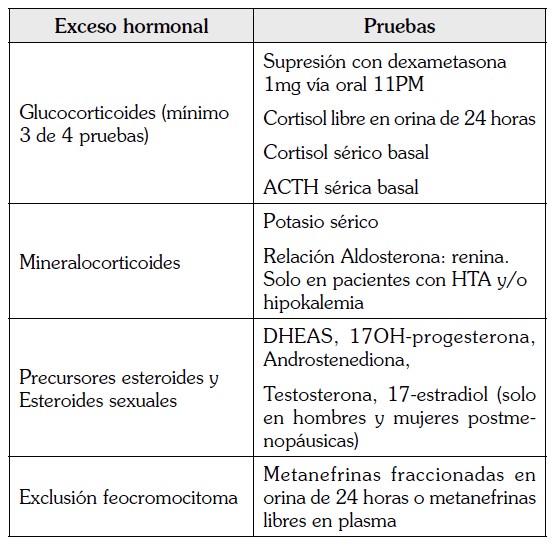
Estudio radiológico
TAC
Las imágenes juegan un papel importante en el diagnóstico diferencial de una masa adrenal, con igual efectividad para TAC y RMN. Típicamente los carcinomas corticoadrenales son tumores no homogéneos, con evidencia de necrosis o hemorragia y márgenes irregulares. Al momento del diagnóstico, el tamaño promedio del tumor es mayor o igual a 10 cm (26).
Los criterios radiológicos se basan en tamaño del tumor y medición de unidades Hounsfield (HU). A mayor tamaño de la masa suprarrenal, la probabilidad de malignidad aumenta; 2% de los "incidentalomas" suprarrenales menores de 4 cm son carcinomas de la corteza suprarrenal y 6% de las lesiones entre 4 y 6 cm, y 25% de las masas suprarrenales incidentales de más de 6 cm son carcinomas. La medición de unidades Hounsfield (HU) en la TAC, ha sido útil para ayudar a diferenciar lesiones benignas de malignas, así: valores de atenuación mayor de 10 HU(unidades Hounsfield) en imágenes sin contraste, mayor de 30 HU en imágenes posteriores al contraste y un "lavado" del contraste (washout) menor de 60% a los 15 minutos, son sugestivas de lesiones malignas (27).
RMN Multiplanar
RMN Multiplanar, separa masas adrenales de estructuras circundantes como hígado o bazo y es extremadamente útil para guiar el abordaje quirúrgico. Los carcinomas corticoadrenales producen típicamente la misma intensidad de señal que el hígado en T1 y presentan un aumento en la intensidad en secuencias T2. Un realce claro después de la administración de gadolinio es seguido por un lavado lento (28).
PET 18-fluorodeoxiglucosa (18F-FDG)
Todos los pacientes con carcinomas corticoadrenales demuestran una captación alta de 18F-FDG, con una relación del valor de captación estandarizado máximo adrenal a hígado mayor de 1.45. En un estudio prospectivo de 77 pacientes operados, la sensibilidad y especificidad para distinguir adenomas de carcinomas corticoadrenales fue de 100% y 88%, respectivamente. Así, en casos indeterminados, 18F-FDG-PET es altamente útil para definir el potencial maligno de un masa adrenal (29).
Sin embargo, ni la TAC, ni la RMN o FDG-PET pueden de manera confiable diferenciar un carcinoma de un feocromocitoma o una metástasis adrenal.
El metomidato está emergiendo como un nuevo radiotrazador. Metomidato se une específicamente a 11-hidroxilasa adrenal y aldosterona sintetaza; por lo tanto, la su captación indica el origen corticoadrenal de una lesión. Metomidato puede ser dado como 11Cmetomidato para PET, o como 123I-iodometomidato para SPECT (30, 31).
Histopatología
El diagnóstico histológico de carcinoma de corteza adrenal no representa gran dificultad cuando el tumor es invasivo; sin embargo, no hay indicadores absolutos de malignidad.
Varios marcadores han sido introducidos para establecer el origen corticoadrenal de masas adrenales.
En 1995, Sasano y colaboradores sugirieron SF1 como un marcador para diferenciar entre tumores de origen corticoadrenal y no corticoadrenal. El valor de este marcador ha sido confirmado en una gran cohorte, en la cual Inmunohistoquímica para SF1 fue positivo en más del 95% de todos los tumores corticoadrenales productores. Además, SF1 brinda información pronostica que es independiente del estadiaje del tumor (32).
Para el diagnóstico de carcinoma versus adenoma, se cuenta con diferentes escalas diagnósticas. La más usada es la de Weiss, con una puntuación de 0 a 9 y se determina de acuerdo a parámetros relacionados con estructura del tumor, citología, evidencia de invasión tumoral y el índice Ki67. Este último además está asociado con disminución en sobrevida global (33).
De suma importancia esta la definición del estado de resección: R0 (resección completa), R1 (tumor residual microscópico) o R2 (tumor residual macroscópico), el cual es un predictor mayor de pronóstico. La violación de la capsula del tumor esta también asociada con recurrencia temprana.
La toma de muestras de tejido para estudio citopatológico (BACAF) no está indicada por el riesgo de complicaciones importantes como hemorragia, ruptura del tumor y riesgo de diseminación, además por el pobre rendimiento diagnóstico en la discriminación entre adenoma y carcinoma suprarrenal (34).
Hay 2 potenciales indicaciones para BACAF (después de excluir el feocromocitoma): primero, para establecer el diagnostico en un tumor adrenal con metástasis, en el cual no se pretende hacer cirugía; segundo, para excluir o demostrar enfermedad metastásica en un paciente con historia de neoplasia maligna extra-adrenal, ya que el resultado puede afectar el tratamiento (1,34).
Estadiaje
El estadio de la enfermedad se gradúa de I a IV, según la clasificación TNM propuesta por ENSAT (European Network for the Study of Adrenal Tumors). Este sistema de estadiaje, define estadios I y II como tumores estrictamente localizados, con un tamaño menor o igual a 5 cm o mayor a 5 cm respectivamente. Tumores estadio III están caracterizados por infiltración a tejidos circundantes, ganglios linfáticos regionales positives o un trombo por el tumor en vena cava y/o renal. El estadio IV se define por la presencia de metástasis distantes. Para detectar enfermedad metastásica temprana, se requieren estudios cuidadosos incluyendo TAC de tórax, previo a la cirugía, ya que la remoción del tumor primario es de valor cuestionable en estadio IV.
La supervivencia para los estadios I, II y III es de 50%, aproximadamente, a los 40 meses y para el estadío IV, de 10% para el mismo tiempo (25).
Estadiaje ENSAT para carcinoma corticoadrenal
Estadío Estadiaje de tumor ENSAT 2008
I T1, NO, MO
II T2, NO, MO
III T1-2, N1, MO
T3-4, NO-1, MO
IV T1-4, NO-1, M1
T1, tamaño tumor ≤5 cm; T2, tamaño de tumor >5 cm; T3, infiltración del tumor a tejidos vecinos; T4, invasión del tumor a órganos adyacentes o trombo tumoral venoso en vena cava o renal. N0, ganglios linfáticos negatives. N1, ganglio(s) linfático(s) positive(s); M0, sin metástasis distantes; M1, presencia de metástasis distantes. Abreviaciones: ENSAT, European Network for the Study of Adrenal Tumors; M, metastasis; N, ganglio linfático; T, tumor.
Tratamiento
Cirugía
Las recomendaciones para el manejo no son absolutas, pues no se dispone hasta el momento de al menos un estudio aleatorizado; en el mejor de los casos se tiene un nivel de evidencia 2, pero casi todas las recomendaciones son niveles de evidencia 3 y 4 (27).
La resección quirúrgica completa es el tratamiento de elección en carcinoma corticoadrenal, ya que es la única opción de lograr la curación. Debe ser realizada idealmente por un cirujano especializado en centros donde se hagan más de 20 adrenalectomías por año (35).
Pacientes con tumor infiltrativo o sospecha de compromiso ganglionar (posible estadio III), debe practicarse adrenalectomía abierta.
Hay evidencia apoyando la hipótesis que carcinomas estadios I y II, podrían ser extraídos por adrenalectomía laparoscópica, aunque esto es materia de controversia (36). Sin embargo este abordaje podría estar justificado en el escenario de incidentalomas no funcionales con evidencia de crecimiento tumoral.
La cirugía está indicada también en el estadio IV, cuando se considera que se puede extirpar el tumor primario y las metástasis, con remoción de más de 90% de la masa tumoral. En algunos pacientes, se requiere una segunda cirugía para extraer todas las lesiones tumorales, como por ejemplo, para resección de metástasis pulmonar (1,2).
Terapia adyuvante
El tratamiento médico adyuvante tiene como objetivo el control del crecimiento tumoral y de cualquier hipersecreción hormonal. El control de la secreción hormonal puede lograrse con fármacos que bloqueen enzimas esteroidogénicas, como ketoconazol, etomidato, metopirona y aminoglutetimida que, a diferencia del mitotane, no poseen efecto sobre la progresión de la enfermedad; por eso se usa preferentemente el mitotane (2, 26,27).
Para los pacientes en estadios I/II llevados a cirugía, no se ha establecido el beneficio del tratamiento adyuvante con mitotane, aunque algunos estudios como el multicéntrico de Terzolo y colaboradores lo sugieren. El estudio demostró que el riesgo de recurrencia y muerte se disminuyó de manera significativa por el uso del mitotane (promedio de sobrevida libre de recurrencia 42 meses en el grupo de mitotane versus 10 and 25 meses en los 2 grupos control; P < 0.01) (38). Derivado de este estudio, algunos centros recomiendan mitotane adyuvante después de la resección completa, aun para R0 (39, 40). Cabe anotar que casi todas las series publicadas tienen un marcado sesgo de selección, como lo demostró un análisis del registro de carcinoma alemán. Este estudio demostró que pacientes que permanecen libres de enfermedad después de la cirugía, rara vez contactan un servicio especializado. Por lo tanto pacientes con riesgo alto de recurrencia son sobrerrepresentados en la mayoría de reportes (41). Así, el pronóstico general de carcinoma corticoadrenal de estadio temprano, podría ser muchísimo mejor que lo reportado y la necesidad de terapia adyuvante no sería obvia, máxime tratándose de un fármaco con efectos adversos importantes. Por todo lo anterior los resultados promisorios del estudio de Terzolo necesitan confirmarse mediante un estudio prospectivo aleatorizado, que se está corriendo actualmente en Italia, ADIUVO (Efficacy of Adjuvant Mitotane Treatment) . el propósito de este estudio es determinar si la terapia adyuvante con mitotane es efectiva en prolongar la sobrevida libre de enfermedad en pacientes con carcinoma corticoadrenal en riesgos de progresión bajo e intermedio, después de resección completa. Actualmente están en la fase de reclutamiento de pacientes y se esperan los resultados para el 2014 (42,43).
En los estadios III y IV, el mitotane permite la regresión tumoral en algunos casos. Sin embargo, es una terapia tóxica que requiere vigilancia de los valores séricos y del manejo por un médico experto en el tema (2,44). Durante el tratamiento, todos los pacientes harán insuficiencia adrenal y tendrán síntomas gastrointestinales, los cuales deben ser tratados prontamente.
En pacientes con resección incompleta (R1) o estado de resección incierto (Rx), debe ofrecerse radioterapia adyuvante en adición al mitotane, para reducir el riesgo alto de recurrencia (1, 45,46).
Enfermedad recurrente
El tratamiento quirúrgico de las recurrencias solo debe realizarse si el tiempo entre la primera operación y la recurrencia es de 6 a 12 meses y si la lesión es susceptible de resección completa, pues solo estos pacientes tienen un periodo clínicamente relevante de sobrevida libre de enfermedad (47). Después de la cirugía, debe iniciarse tan pronto como sea posible, la terapia adyuvante. En pacientes con enfermedad recurrente a pesar de terapia con mitotane, se debe considerar la adición de otro fármaco como estreptozotocina o radioterapia para recurrencia local (48).
Enfermedad avanzada
En enfermedad avanzada, la cirugía citorreductora es benéfica únicamente en pacientes con exceso severo de hormonas que no puedan controlarse de otra manera.
La terapia médica debe instaurase tan pronto como se haga el diagnostico. Hasta el momento, los regímenes de primera línea son monoterapia con mitotane; etoposido, doxorrubicina, cisplatino más mitotane (49); o streptozotocina mas mitotane (48). El tratamiento se elige según el criterio médico y la velocidad de progresión del tumor. Para monoterapia con mitotane se puede seguir el siguiente esquema: después de su inicio, la dosis se aumenta en pocos días hasta 6 gramos/día y se obtienen niveles séricos después de 3 semanas de tratamiento. Si la concentración sérica del mitotane es mayor de 7 mg/L, se continua monoterapia con mitotane, si los niveles séricos son inferiores a 7 mg/L, se considera la adición inmediata de otros fármacos citotóxicos (1).
En pacientes con progresión tumoral a pesar de terapia médica de primera línea, son considerados candidatos a regímenes alternos (por ejemplo capecitabina mas gemcitabina). (50).
Si el mitotane solo no es capaz de controlar el exceso hormonal, deben adicionarse otros fármacos adrenostáticos, tales como ketoconazol, metopirona, etomidato o mifepristona (RU486) (51, 52). Se están probando terapias más nuevas; resultados preliminares de ensayos fase I con un inhibidor oral del receptor IGF-1 (OSI-906) son promisorios. En 5 de 16 pacientes, se logró estabilización de la progresión del carcinoma, durante más de 12 semanas, incluyendo un paciente con una reducción de más del 80% de la masa tumoral. Un ensayo fase III de este medicamento, como terapia de segunda o tercera línea se está corriendo en varios países (53).
Se ha usado radioterapia paliativa en lesiones metastásicas sintomáticas (ver tabla de radioterapia).
Ablación con radiofrecuencia es una alternativa para metástasis <5 cm (54).
Seguimiento
El seguimiento de los pacientes con carcinoma de corteza suprarrenal se hace con marcadores endocrinos cada 3 meses si la resección fue completa y según el perfil prequirúrgico, para evaluar recaídas; en pacientes con resección tumoral incompleta, debe iniciarse el tratamiento para mantener las hormonas suprarrenales dentro de límites normales.
Durante el primer año de seguimiento, se requiere evaluación con tomografía toracoabdominal cada 3 meses, luego cada 3 a 6 meses por 5 años y, luego, anualmente.
En pacientes sin evidencia de enfermedad, se recomienda el seguimiento durante mínimo de 10 años después de la cirugía. (1, 2,27).
Pronóstico
El pronóstico general de carcinoma corticoadrenal es pobre, con una sobrevida a 5 años que varía entre 37% y 47% (55). Además del grado de resección quirúrgica, el estadio del tumor al momento del diagnóstico, permanece como el factor pronóstico más relevante. Sin embargo la sobrevida difiere ampliamente para cualquier estadío del tumor, reflejando la heterogeneidad de la enfermedad. Desafortunadamente, se disponen de datos muy limitados sobre marcadores clínicos, histológicos y moleculares para predicción de comportamiento del tumor.
Conclusiones
A pesar de la baja prevalencia del carcinoma corticoadrenal, lo cual dificulta su estudio, en los últimos años se han hecho grandes avances en la comprensión de su biología, patogenia y el concurso de diferentes alteraciones genéticas. Esto ha abierto el camino para nuevas terapias farmacológicas, como es el inhibidor del receptor IGF1. También ha posibilitado un mejor estadiaje del tumor y la promesa a futuro de marcadores tumorales que mejoren el rendimiento diagnóstico y pronóstico.
El carcinoma de nuestra paciente, se sitúa en el 15% de estos tumores diagnosticados como incidentalomas. A pesar de su gran tamaño, no había hecho clínica de compresión para sospecharlo ni síndrome de exceso hormonal. Es un estadio II (T2N0M0), en la cual está indicada la adrenalectomía abierta, la cual se realizó. La radioterapia queda a criterio individual, y en este caso se administró por la invasión microscópica de vasos sanguíneos que presentaba. No sabemos en este momento si se beneficiaría o no de la terapia adyuvante con mitotane y este interrogante será despejado en el estudio ADIUVO.
Bibliografía
1. Fassnacht, M., Libé, R., Kroiss, M., Allolio, B. Adrenocortical carcinoma: a clinician's update. Nat. Rev. Endocrinol. 7, 323-335 (2011). [ Links ]
2. Pabon, N., Gomez, C., Garavito, G., Jimenez, C. Carcinoma corticosuprarrenal: estado actual de nuestro conocimiento. Rev Colomb Cancerol 2005;9(3):123-129. [ Links ]
3. Universität Würzburg Offizielle Homepage des Deutsche Nebennierenkarzinom-Registers [online], http://www.nebennierenkarzinom.de/ (2010). [ Links ]
4. Libe, R., Bertherat, J. Molecular genetics of adrenocortical tumours, from familial to sporadic diseases.Eur J Endocrinol 2005;153:477-87. [ Links ]
5. Geller, J., Mertens, R., Weiss, L. Adrenocortical carcinoma many questions remain unanswered. The Endocrinologist 2005;15:3009-312. [ Links ]
6. Ribeiro, R., Pinto, E., Zambetti, G. Familial predisposition to adrenocortical tumors: Clinical and biological features and management strategies. Best Practice & Research Clinical Endocrinology & Metabolism 24 (2010) 477e490. [ Links ]
7. Tissier, F., Cavard, C., Groussin, L et al. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Research 2005; 65: 7622e7627. [ Links ]
8. Bisgaard, M., Fenger, K., Bulow S et al. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Human Mutation 1994; 3: 121e125. [ Links ]
9. DeChiara, T., Robertson EJ & Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 1991; 64: 849e859. [ Links ]
10. Lapunzina, B., del Campo, C., Delicado, N.et al. Clinical guide to the management of patients with BeckwitheWiedemann syndrome. Anales de Pediatria (Barcelona) 2006; 64: 252e259. [ Links ]
11. Teh SH & Ong GB. Early presentation of right adrenal mass, hepatoblastoma and hepatic cavernous haemangioma in BeckwitheWiedemann Syndrome. The Medical Journal of Malaysia 2007; 62: 345e346. [ Links ]
12. Schimmer, B. & White, P. Minireview: steroidogenic factor 1: its roles in differentiation, development, and disease. Mol. Endocrinol. 24, 1322-1337 (2010). [ Links ]
13. Sbiera, S. et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J. Clin. Endocrinol. Metab. 95, E161-E171 (2010). [ Links ]
14. Bernini, G. et al. Angiogenesis in human normal and pathologic adrenal cortex. J. Clin. Endocrinol. Metab. 87, 4961-4965 (2002). [ Links ]
15. Isobe, M., Bravo, E., Givol, D et al. Localization of gene for human p53 tumour antigen to band 17p13. Nature 1986; 320: 84e85. [ Links ]
16. Bertherat, J., Gimenez, R. New insights in the genetics of adrenocortical tumours, pheoch-romocytomas and paragangliomas. Horm Metab Res 2005;37:384-90. [ Links ]
17. Sidhu, S., Gicquel, C., Bambach C et al. Clinical and molecular aspects of adrenocortical tumourigenesis. ANZ J Surg 2003;73:727-38. [ Links ]
18. Zambetti, G. The p53 mutation "gradient effect" and its clinical implications. Journal of Cellular Physiology 2007; 213: 370e373. [ Links ]
19. Gonzalez, K., Noltner, K., Buzin, C et al. Beyond LieFraumeni Syndrome: clinical characteristics of families with p53 germline mutations. Journal of Clinical Oncology 2009; 27: 1250e1256. [ Links ]
20. Beuschlein, F., Fassnacht, M., Klink, A., Allolio, B. & Reincke, M. ACTH-receptor expression, regulation and role in adrenocortial tumor formation. Eur. J. Endocrinol. 144, 199-206 (2001). [ Links ]
21. Stewart, P. M., Walker, B. R., Holder, G., O'Halloran, D. & Shackleton, C. H. 11 beta-Hydroxysteroid dehydrogenase activity in Cushing's syndrome: explaining the mineralocorticoid excess state of the ectopic adrenocorticotropin syndrome. J. Clin. Endocrinol. Metab. 80, 3617-3620 (1995). [ Links ]
22. Universität Würzburg Offizielle Homepage des Deutsche NebennierenkarzinomRegisters [online], http://www.nebennierenkarzinom.de/ (2010). [ Links ]
23. Hyodo, T., Megyesi, K., Kahn, C. R., McLean, J. P. & Friesen, H. G. Adrenocortical carcinoma and hypoglycemia: evidence for production of nonsuppressible insulin-like activity by the tumor. J. Clin. Endocrinol. Metab. 44, 1175-1184 (1977). [ Links ]
24. Francois C, Rangachari B, Bova D. Case report: mammography and sonography of pathologically proven adrenal cortical carcinoma metastasic to the breast. Am J Roentgenol 2005;184:1279-81. [ Links ]
25. European Network for the Study of Adrenal Tumours Adrenocortical carcinomas [online], http://www.ensat.org/acc.htm (2010). [ Links ]
26. Fassnacht, M. & Allolio, B. Clinical management of adrenocortical carcinoma. Best Pract. Res. Clin. Endocrinol. Metab. 23, 273-289 (2009). [ Links ]
27. Schteingart, D., Doherty, G., Gauger, P. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocrine Related Cancer 2005;12:667-80. [ Links ]
28. Ilias, I., Sahdev, A., Reznek, R. H., Grossman, A. B. & Pacak, K. The optimal imaging of adrenal tumours: a comparison of different methods. Endocr. Relat. Cancer 14, 587-599 (2007). [ Links ]
29. Groussin, L. et al. 18F-Fluorodeoxyglucose positron emission tomography for the diagnosis of adrenocortical tumors: a prospective study in 77 operated patients. J. Clin. Endocrinol. Metab. 94, 1713-1722 (2009). [ Links ]
30. Hahner, S. et al. [123 I]Iodometomidate for molecular imaging of adrenocortical cytochrome. [ Links ]
31. Hennings, J. et al. [11C]metomidate positron emission tomography of adrenocortical tumors in correlation with histopathological findings. J. Clin. Endocrinol. Metab. 91, 1410-1414 (2006). [ Links ]
32. Sasano, H. et al. Transcription factor adrenal 4 binding protein as a marker of adrenocortical malignancy. Hum. Pathol. 26, 1154-1156 (1995). [ Links ]
33. Aiba M, Fujibayashi M. Histopathological diagnosis and prognostic factors in adrenocortical carcinoma. Endocr Pathol 2005;16:13-22. [ Links ]
34. Quayle, F. J. et al. Needle biopsy of incidentally discovered adrenal masses is rarely informative and potentially hazardous. Surgery 142, 497-502 (2007). [ Links ]
35. Murphy, M. M. et al. Trends in adrenalectomy: a recent national review. Surg. Endosc. 24, 2518-2526 (2010). [ Links ]
36. Porpiglia, F. et al. Retrospective evaluation of the outcome of open versus laparoscopic adrenalectomy for stage I and II adrenocortical cancer. Eur. Urol. 57, 873-878 (2010). [ Links ]
37. Brix, D. et al. Laparoscopic versus open adrenalectomy for adrenocortical carcinoma: surgical and oncologic outcome in 152 patients. Eur. Urol. 58, 609-615 (2010). [ Links ]
38. Terzolo, M. et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N. Engl. J. Med. 356, 2372-2380 (2007). [ Links ]
39. Huang, H. & Fojo, T. Adjuvant mitotane for adrenocortical cancer-a recurring controversy. J. Clin. Endocrinol. Metab. 93, 3730-3732 (2008). [ Links ]
40. Terzolo, M., Fassnacht, M., Ciccone, G., Allolio, B. & Berruti, A. Adjuvant mitotane for adrenocortical cancer-working through uncertainty. J. Clin. Endocrinol. Metab. 94, 1879-1880 (2009). [ Links ]
41. Fassnacht, M. et al. Improved survival in patients with stage II adrenocortical carcinoma followed up prospectively by specialized centers. J. Clin. Endocrinol. Metab. 95, 4925-4932 (2010). [ Links ]
42. Terzolo, M., Fassnacht, M., Ciccone, G., Baudin, E., Haak, H., Berruti, A. Efficacy of Adjuvant Mitotane Treatment in Prolonging Recurrence-free Survival in Patients With Adrenocortical Carcinoma at Low-intermediate Risk of Recurrence. 2008-2014 [ Links ]
43. US National Institutes of Health Clinicaltrials.gov [online], http://clinicaltrials.gov/ct2/show/NCT00777244 (2010). [ Links ]
44. Daffara, F. et al. Prospective evaluation of mitotane toxicity in adrenocortical cancer patients treated adjuvantly. Endocr. Relat. Cancer 15, 1043-1053 (2008). [ Links ]
45. Polat, B. et al. Radiotherapy in adrenocortical carcinoma. Cancer 115, 2816-2823 (2009). [ Links ]
46. Sabolch, A. et al. Adjuvant and definitive radiotherapy for adrenocortical carcinoma. Int. J. Radiat. Oncol. Biol. Phys. doi:10.1016/j.ijrobp.2010.04.030. [ Links ]
47. Erdogan, I. et al. Impact of surgery on clinical outcome in patients with recurrence of adrenocortical carcinoma. Endocrine Abstracts (10th European Congress of Endocrinology), 20, P194 (2009). [ Links ]
48. Khan, T. S. et al. Streptozocin and o,p'DDD in the treatment of adrenocortical cancer patients: long-term survival in its adjuvant use. Ann. Oncol. 11, 1281-1287 (2000). [ Links ]
49. Berruti, A. et al. Etoposide, doxorubicin and cisplatin plus mitotane in the treatment of advanced adrenocortical carcinoma: a large prospective phase II trial. Endocr. Relat. Cancer 12, 657-666 (2005). [ Links ]
50. Sperone, P. et al. Gemcitabine plus metronomic 5-fluorouracil or capecitabine as a second-/third-line chemotherapy in advanced adrenocortical carcinoma: a multicenter phase II study. Endocr. Relat. Cancer 17, 445-453 (2010). [ Links ]
51. Castinetti, F. et al. Merits and pitfalls of mifepristone in Cushing's syndrome. Eur. J. Endocrinol. 160, 1003-1010 (2009). [ Links ]
52. Johanssen, S. & Allolio, B. Mifepristone (RU 486) in Cushing's syndrome. Eur. J. Endocrinol. 157, 561-569 (2007). [ Links ]
53. Carden, C. P. et al. Phase I study of intermittent dosing of OSI-906, a dual tyrosine kinase inhibitor of insulin-like growth factor-1 receptor (IGF-1R) and insulin receptor (IR) in patients with advanced solid tumors. J. Clin. Oncol. 28 (Suppl.), abstr 2530 (2010). [ Links ]
54. Bauditz, J., Quinkler, M. & Wermke, W. Radiofrequency thermal ablation of hepatic metastases of adrenocortical cancer-a case report and review of the literature. Exp. Clin. Endocrinol. Diabetes 117, 316-319 (2009). [ Links ]
55. Bilimoria, K. Y. et al. Adrenocortical carcinoma in the United States: treatment utilization and prognostic factors. Cancer 113, 3130-3136 (2008). [ Links ]

