










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Med
Print version ISSN 0121-5256
rev.fac.med vol.20 no.2 Bogotá July/Dec. 2012
ARTÍCULO DE REVISIÓN
ESTADO DEL ARTE: TUMORES DE CRESTA NEURAL, NEUROBLASTOMA FISIOPATOLOGÍA, DIAGNÓSTICO Y TRATAMIENTO
STATE OF ART: NEURAL CREST TUMORS, NEUROBLASTOMA (NB) PATHOPHYSIOLOGY, DIAGNOSIS AND TREATMENT
ESTADO DE ARTE: TUMORES DE CRESTA NEURAL, NEUROBLASTOMA FISIOPATOLOGIA, DIAGNÓSTICO E TRATAMENTO
ANDRÉS M. ÁLVAREZ, MD SPECa*,REYNALD LAMARRE, MDb*
a General Medicine and Surgery, Specialist in Bioethics Universidad El Bosque Research Fellow Orthopaedic and Neurological Surgery Department Cleveland Clinic, USA
b Ross University School of Medicine Research Fellow Orthopaedic Department Cleveland Clinic, USA.
*Correspondencia: Andrés M. Álvarez MD. Correo electrónico: andresmauricioalvarez07@gmail.com Reynald Lamarre, M.D. Correo electrónico: reynald.lamarre@gmail.com
Recibido: Agosto 30 de 2012 Aceptado: Noviembre 26 de 2012
Resumen
El neuroblastoma es el tumor sólido extracraneal más frecuente en la infancia, con incidencia de 650 casos al año en los Estados Unidos de America y 100 casos por año en Italia y España. Se trata de un tumor que evoluciona de forma variable, desde la regresión espontánea hasta un comportamiento muy maligno, sobre todo en niños en edades mayores y con la existencia de enfermedad diseminada. Los signos y síntomas de presentación del neuroblastoma reflejan la localización del tumor y debido a las múltiples presentaciones clínicas, puede confundirse con gran variedad de patologías. Su tratamiento incluye la cirugía, la quimioterapia, la radioterapia y la terapia inmunológica. El papel de cada uno se determina anticipando el comportamiento clínico del tumor en cada caso, considerando la edad, el estadio y determinados parámetros biológicos. En los últimos 10 años, la estratificación por grupos de riesgo de los pacientes con neuroblastoma, basada en el análisis de un panel amplio de variables clínicas y biológicas, entre las que destacan la edad, el estadio y la amplificación del N-MYC, ha permitido un diagnóstico temprano y nuevas pautas para una mejoría importante en su tratamiento. El tratamiento del neuroblastoma se diseña hoy en día según grupos de riesgo, que se definen por parámetros clínicos y biológicos cada vez más sofisticados. El mejor conocimiento de la biología del neuroblastoma ha permitido distinguir entre aquellos tumores que pueden curarse con un tratamiento mínimo y los que requieren de un tratamiento multidisciplinario y complejo para tener posibilidades de curación. Es fundamental realizar al diagnóstico temprano con indicaciones de estadio y desarrollo de una terapia oportuna.
Palabras clave: Neuroblastoma, cresta neural, tumor cerebral, infancia, N-MYC, médula, síndrome de Homer, NCMAM.
Abstract
Neuroblastoma (NB) is the most common extracranial solid cancer in childhood and the most common cancer in infancy, with an annual incidence of about 650 cases per year in the US, and 100 cases per year in Italy and Spain. Roughly 50 percent of neuroblastoma cases occur in children younger than two years of age. It is a neuroendocrine tumor, arising from neural crest cells of the sympathetic nervous system (SNS). It most frequently originates in one of the adrenal glands, but can also develop in nerve tissues located in the neck, chest, abdomen, and pelvis. It is a highly variable tumor that can spontaneously regress or have highly malignant behavior, especially in older children with disseminated disease. Signs and symptoms of neuroblastoma reflect the presentation of tumor location and because of the multiple clinical presentations, can be confused with a variety of pathologies. Treatment modalities include surgery, chemotherapy, radiotherapy and immunotherapy. The role of each method is determined in advance of the clinical behavior of the tumor on a case specific basis, consideration of the age, stage and biological parameters. Over the past decade, stratification of patients with risk factors for neuroblastoma based on the analysis of a large panel of clinical and biological variables, among which are age, stage and N-MYC amplification, has allowed for early diagnosis and the generation of new guidelines for major advancements in treatment. Today, treatment of neuroblastoma has become increasingly sophisticated and is influenced by risk factors as well as clinical and biological parameters. A better understanding of the biology of neuroblastoma has allowed for the differentiation between tumors that can be cured with minimal treatment and those requiring a multidisciplinary and complex approach in order to have a chance of cure. Early diagnosis with staging is essential in the development of an appropriate therapy.Key words:Neuroblastoma, neural crest, brain tumor, childhood, N-MYC, bone, Homer syndrome, NCMAM.
Resumo
O neuroblastoma é o tumor sólido extra-craneal mais frequente na infância, com incidência de 650 casos por ano nos Estados Unidos da América e 100 casos por ano na Itália e Espanha. Trata-se de um tumor que evolui de forma variável, desde a regressão espontânea até um comportamento muito maligno, sobretudo em crianças com idades maiores e com a existência de doença disseminada. Os sinais e sintomas de apresentação do neuroblastoma refletem a localização do tumor e devido as múltiplas apresentações clínicas, pode ser confundido com uma grande variedade de patologias. Seu tratamento inclui a cirurgia, a quimioterapia, a radioterapia e a terapia imunológica. O papel de cada um é determinado antecipando o comportamento clínico do tumor em cada caso, considerando a idade, o estádio e determinados parâmetros biológicos. Nos últimos 10 anos, a estratificação por grupos de risco dos pacientes com neuroblastoma, baseada na análise de um painel amplo de variáveis clínicas e biológicas, entre as que destacam a idade, o estádio, e a amplificação do N-MYC, permitem um diagnostico precoce e novas pautas para uma melhoria importante no seu tratamento. O tratamento do neuroblastoma é feito hoje em dia de acordo com grupos de risco, que são definidos por parâmetros clínicos e biológicos cada vez mais sofisticados. O melhor conhecimento da biologia do neuroblastoma permitiu distinguir entre os tumores que podem ser curados com um tratamento mínimo e os que requerem um tratamento multidisciplinar e complexo para ter possibilidades de cura. É fundamental realizar o diagnóstico precoce com indicações de estádio e desenvolvimento de uma terapia oportuna.
Palavraschave: Neuroblastoma, cresta neural, tumor cerebral, infância, N-MYC, medula, síndrome de Homer, NCMAM.
Introducción
El neuroblastoma es el tumor sólido extracraneal más frecuente en la infancia. Es un tumor derivado de las células precursoras de la médula adrenal y del sistema nervioso simpático (1,2). Su comportamiento biológico a lo largo de los tiempos parecía impredecible, unas veces muy agresivo y resistente al tratamiento y en otras ocasiones, curable incluso sin tratamiento. El neuroblastoma puede desaparecer espontáneamente en el lactante, o madurar y convertirse en un ganglioneuroblastoma benigno (1, 3). Sin embargo, en la mayoría de los niños mayores de un año, el tumor está diseminado y tiene un pronóstico muy desfavorable. La aplicación de técnicas de genética molecular en el diagnóstico del tumor y el estudio de su patogenia ha permitido distinguir dos tipos de neuroblastoma con unas características clínicas y pronóstico muy diferente desde el inicio de la enfermedad, detectando también la posibilidad de que exista un tercer tipo de neuroblastoma que resulte de la evolución del primero al segundo (1, 4).
Epidemiología
El neuroblastoma es el tumor sólido extracraneal más frecuente en la infancia, representa entre el 8 y el 10% de todos los tumores pediátricos, en Colombia no se encontraron estudios de esta patología pero la incidencia en Sur America es de 9,1 x 106 habitantes, similar a la de otros países, por ejemplo España e Italia (2-4). Se trata de una neoplasia prácticamente exclusiva de la infancia, la edad media en el momento del diagnóstico es de 17,3 meses. El 40,1% de los niños se diagnostica antes del año de edad, el 89,4% antes de los 4 años y el 97,8% antes de los 10 años. No se ha descrito una asociación clara entre ningún factor ambiental, químico, irradiación o biológico y el desarrollo del neuroblastoma. Los paises con mayores estudios y diagnósticos tempranos se encuentran en el norte de Europa, considerando su notable aumento en los ultimos años (1, 5).
Diferenciación Anatomopatológica
El neuroblastoma típico se caracteriza microscópicamente por la presencia de nidos de células tumorales, separados por septos fibrovasculares con áreas de necrosis y calcificación (2, 5, 6). Las células tumorales son de morfologia redonda, pequeña y con núcleo hipercromático denso y poco citoplasma (neuroblastos) (figura 1). Los neuroblastomas más primitivos muestran neuropilos o procesos neuríticos, además, es característica la presencia de rosetas de Homer-Wright, compuestas de neuroblastos rodeando áreas de neurópilos eosinofílicos. El ganglioneuroma está compuesto por células ganglionares, neurópilos y células de Schwann. Los ganglioneuroblastomas son un grupo heterogéneo de tumores con áreas de neuroblastoma y otras de galioneuroma (2,7). Cuando la clínica y la anatomía patológica no son concluyentes, deben utilizarse métodos inmunohistoquímicos y de biología molecular para hacer el diagnóstico diferencial con otros tipos de tumores, anticuerpos monoclonales que incluyen la enolasa (NSE), sinaptofisina, gangliósido GD2, molécula de adhesión de la célula neural (NCMAM), tirosín cinasa (TRK) y cromogranina A (CGA) (1, 2, 8).
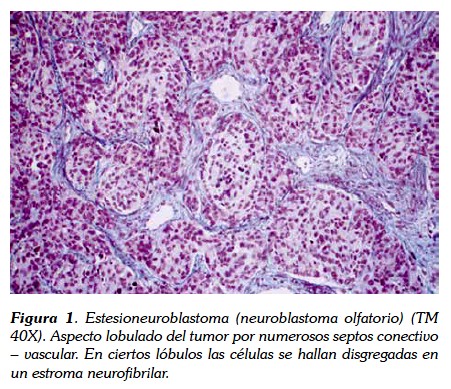
Durante años, se ha utilizado la clasificación de Shimada del Childrens Cancer Group (CCG), que relaciona la edad del paciente con datos histológicos de maduración tumoral. Clasifica los neuroblastomas en ricos o pobres en estroma, según la presencia o ausencia de células de Schwann, el grado de diferenciación y el índice de mitosis-cariorrexis. Los tumores nodulares con estroma abundante y los indiferenciados pobres en estroma tienen un mal pronóstico (8).
Clínica
La clínica del neuroblastoma depende del origen y extensión del tumor primario. El 80% de los niños mayores y el 40% de los lactantes se diagnostican cuando el tumor está diseminado. La diseminación metastásica se realiza por vía hematógena y linfática. La diseminación por vía sanguínea es más frecuente a la médula ósea, hueso, hígado y piel. La diseminación linfática ocurre en el 35% de los casos (1,5,9,10). Cuando existen adenopatías infiltradas fuera de la cavidad de origen, la enfermedad está ya diseminada; sin embargo, la diseminación linfática exclusiva tiene mejor pronóstico que la diseminación hematógena. La mayoría son de localización abdominal (65%), la localización adrenal es ligeramente más alta en niños (40%) que en lactantes (25%). En lactantes, se ven más tumores cervicales y torácicos (11,12).
Aproximadamente, en el 1% de los pacientes el tumor primario no se localiza, los signos y síntomas del neuroblastoma reflejan la localización del tumor primario, la afectación regional y la enfermedad metastásica. La enfermedad abdominal puede producir sensación de plenitud, dolor y distensión, siendo rara la obstrucción intestinal o puede permanecer asintomática diagnosticándose en una exploración de rutina, donde se palpa una masa fija y dura. Cuando el tumor se localiza en el órgano de Zuckerland, puede dar lugar a síntomas compresivos vesicales o intestinales (molestias urinarias, estreñimiento) (11,13,14).
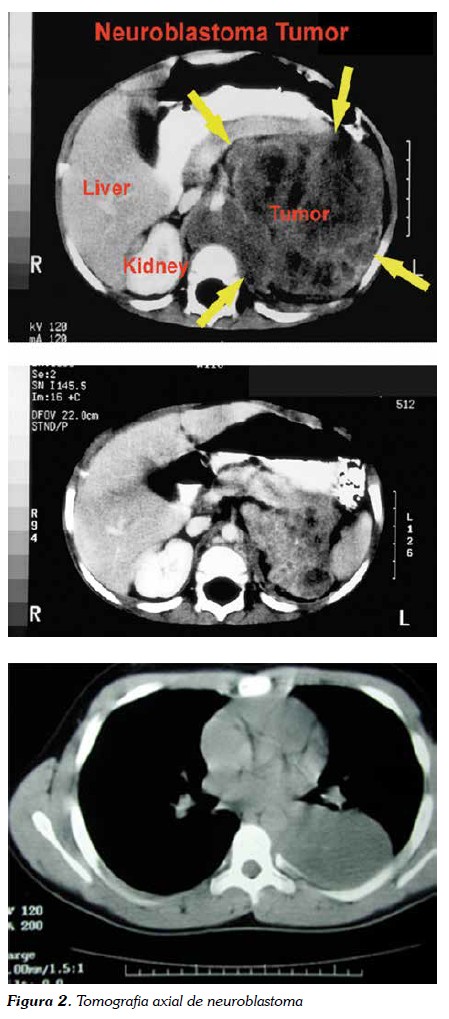
En algunas ocasiones, el tamaño del tumor primario o de las metástasis abdominales provoca una compresión venosa y del drenaje linfático de las extremidades inferiores, apareciendo edema en éstas y en el escroto (14-16). Los tumores primarios torácicos a menudo son diagnosticados de manera casual, mediante radiografías de tórax solicitadas para evaluar traumatismos o infecciones. En esta localización, la sintomatología puede ser nula o pueden presentarse como un cuadro de insuficiencia respiratoria superior o tos crónica (figura 2) (2, 5). Ocasionalmente, la masa es tan grande que da lugar a un síndrome de la vena cava superior, que constituye una urgencia oncológica. Las masas torácicas altas o cervicales pueden producir un síndrome de Horner (ptosis, miosis y enoftalmos) o una heterocromía del iris (15,16).
Los tumores paraespinales, torácicos, abdominales o pélvicos, pueden extenderse por los forámenes neurales de los cuerpos vertebrales y producir síntomas compresivos, desde dolor radicular y síndrome de compresión medular, hasta disfunciones vesicales o intestinales. El neuroblastoma diseminado puede presentarse con síntomas inespecíficos, como: fiebre, irritabilidad, pérdida de peso, etc. (16,17). Las metástasis óseas se manifiestan por dolor. La infiltración de la médula ósea da lugar a anemia con presencia de eritroblastos en sangre periférica, leucopenia y trombopenia con o sin síndrome hemorrágico asociado.
La infiltración tumoral del tejido retroorbitario produce un signo clínico característico: "los ojos de oso panda", con protopsis y equímosis periorbitales (1,2,18). En el lactante, es característica la presentación de un neuroblastoma suprarrenal con diseminación cutánea, que se caracteriza por un número variable de nódulos subcutáneos azulados que se blanquean por la presión en la médula ósea y el hígado . Estos neuroblastomas se clasifican en un estadio 4s (5,8,17). Se trata de una presentación multicéntrica más que de metástasis a distancia, en el 70% afecta a lactantes menores de 3 meses y tiene un pronóstico muy bueno, ya que se curan más del 80%, unas veces con tratamiento y otras porque el tumor regresa espontáneamente (19). La clínica y la evolución dependen más del grado de infiltración hepática que del tamaño y localización del tumor, a veces, la hepatomegalia es tan masiva que produce un cuadro de insuficiencia respiratoria. Se ha visto que en los dos primeros meses de vida el pronóstico es peor, porque el tumor puede crecer muy deprisa y provocar complicaciones importantes antes de iniciar su regresión (20,21).
Síndromes paraneoplásicos
Existen bastantes síndromes paraneoplásicos que se pueden asociar, tanto a neuroblastoma localizado como diseminado (1,2,22,23). El 4% de los neuroblastomas se asocian a una encefalopatía mioclónica que se manifiesta por opsoclonus (movimientos oculares rápidos multidireccionales), mioclonías y ataxia truncal. Estos enfermos tienen buen pronóstico en cuanto al neuroblastoma, pero la sintomatología neurológica puede persistir a pesar de haberse curado el tumor (24-26). Se cree que se debe a anticuerpos dirigidos contra el tumor, pero con acción cruzada con células nerviosas del cerebelo y cerebro del enfermo. Estos niños mejoran con tratamiento con corticoides además de quimioterapia. El riesgo de secuelas neurológicas a largo plazo es elevado; por lo que, teniendo en cuenta el buen pronóstico del tumor que las origina, deben considerarse a la hora de planificar el tratamiento (27,28).
Otro síndrome paraneoplásico asociado al neuroblastoma consiste en una diarrea secretora con hipocaliemia y deshidratación secundaria a la secreción tumoral de VIP (vasopéptido intestinal) y que se observa en el 7-9% de los neuroblastomas (29,30). Los tumores secretores de VIP son histológicamente más benignos (ganga oneuromas y ganglioneuroblastomas) y tienen un mejor pronóstico, la diarrea cede con la resección del tumor. Recientemente, se ha comprobado que estos tumores, además de VIP, secretan somatostatina y que ambas sustancias inducen la diferenciación del tumor in vitro e in vivo (1,3,31).
Finalmente, aunque es raro, algunos tumores se asocian a hipercalcemia cuyo origen aún no ha sido esclarecido, pero podría deberse a la producción tumoral de una sustancia parecida a la paratohormona (32).
Diagnóstico
Existen criterios establecidos para el diagnóstico, estadiaje y valoración de la respuesta al tratamiento del neuroblastoma, distinguidos según la caracterización del inicio de la patología, y distinción anatomo histológica (2,33). El diagnóstico del neuroblastoma se puede realizar hoy en día con relativa facilidad, utilizando métodos diagnósticos e histológicos. En 1986, un grupo de expertos estableció una serie de criterios para el diagnóstico, anatomía patológica (IPCN), estadiaje (INSS) y valoración de la respuesta al tratamiento (INRC) del neuroblastoma. (Figura 3) (34-36). Para el diagnóstico del neuroblastoma se debe cumplir con uno de estos criterios:
• Existe un diagnóstico inequívoco anatomopatológico con o sin aumento de excreción urinaria de catecolaminas fraccionadas o sus metabolitos.
• Existe un aspirado o biopsia de médula ósea positiva, para células tumorales con positividad simultánea de las catecolaminas o sus metabolitos en orina o suero. Desde el punto de vista inmunocitoquímico, se recomienda la utilización de al menos, dos de los siguientes anticuerpos monoclonales: NSE, sinaptofisina y CGA (1,32,35-37).
La obtención de tejido para el diagnóstico debe realizarse por cirugía/biopsia o mediante biopsia percutánea. La punción aspiración con aguja fina (PAAF) tiene el inconveniente de que el material obtenido puede ser insuficiente, la indicación más aceptada es en el diagnóstico de las recidivas (38). Es fundamental asegurar suficiente material para el diagnóstico, sobre todo en los casos dudosos. El estudio genético del tumor es imprescindible para conocer el pronóstico y planificar el tratamiento, por lo tanto, la biopsia diagnóstica debe hacerse siempre en un centro en el que se asegure el correcto manejo de las muestras, así como su envío a los centros de referencia (39-41).
Más del 90% de los pacientes excretan niveles altos de catecolaminas y sus metabolitos en orina, debido a que las células tumorales carecen de metiltransferasas, una de las enzimas que metabolizan la dopamina (42-44). La determinación de catecolaminas en orina, continúa siendo uno de los métodos más fiables y baratos para el seguimiento del tumor durante y al finalizar el tratamiento (2,45). Actualmente, esta determinación se realiza en muestra de orina espontánea y se mide en mg/g de creatinina excretada en orina. Para que los valores sean positivos, tienen que ser superiores a 3 DS respecto a los valores normales para la edad del niño. Cuando las catecolaminas en orina son negativas, debe determinarse la dopamina sérica o urinaria para identificar tumores indiferenciados secretores únicamente de dopamina. Por último, el 10% de los neuroblastomas no produce catecolaminas (46,47).
El estudio del tumor primario se realiza según su localización. En los torácicos el diagnóstico inicial se realiza por Rx de tórax, y en los abdominales con una ecografía abdominal. Además, es imprescindible la realización de un TAC o una RM para valorar la extensión del tumor primario, la RM es el método de elección para estudiar los tumores paravertebrales, que pueden meterse por los agujeros de conjunción y afectar a la médula espinal. En los tumores torácicos inferiores del lado izquierdo, hay que valorar la extensión entre T9 y T12, la infiltración a través de los forámenes de conjunción y la infiltración de la aorta descendente, porque existe riesgo de que la arteria medular esté afectada (48-50), debe realizarse una gammagrafía con I-123 MIBG (yodometilbencilguanidina), que nos dará una imagen del tumor primario y de la presencia de metástasis. Cuando esta prueba es positiva al diagnóstico, supone una herramienta muy útil para el seguimiento de la enfermedad durante y al finalizar el tratamiento (51-53). Si el tumor no capta MIBG, se debe hacer una gammagrafía ósea con Tc-99 para evaluar la presencia de metástasis óseas. El diagnóstico del neuroblastoma basado en datos radiológicos o gammagráficos es insuficiente; ya que, tumores benignos, como el ganglioneuroma, feocromocitoma y otros tumores neurales pueden ser difíciles de distinguir, pueden ser secretores de catecolaminas y pueden captar I-123 MIBG (54-56).
Evaluación paraclínica
Existen algunas pruebas diagnósticas que se consideran indispensables para definir el estadio del neuroblastoma, éstas deben seguir criterios uniformes para facilitar la comparabilidad (19,57). El estudio de extensión del neuroblastoma debe incluir (1,2,57,58):
• Dos muestras de aspirado y biopsia de médula ósea realizadas en ambas crestas ilíacas. En caso de no poder realizarse la biopsia, se realizarán cuatro aspirados, basta que sea positiva una de las muestras para el diagnóstico de afectación medular, se excluye la afectación medular si las cuatro muestras son negativas. El estudio morfológico debe complementarse con estudios inmunohistoquímicos y de biología molecular. En la mayoría de los casos, si existe afectación medular, ésta es evidente, se ven células tumorales aisladas o en grupo formando rosetas.
• Las metástasis óseas se detectan en la gammagrafía con I-123 MIBG, pero en el caso de que el tumor no capte, debe realizarse una gammagrafía ósea con Tc-99. Si existe captación de Tc-99 en algún hueso, hay que completar el estudio con Rx/TAC del hueso. En los lactantes en los que exista captación ósea de Tc-99 pero la Rx/TAC del hueso no muestre afectación ósea, no se considerará la existencia de metástasis óseas.
• La enfermedad pleuropulmonar es infrecuente y se estudia con Rx de tórax. Se realizará un TAC de tórax si existen imágenes sospechosas en la Rx.
• El estudio de adenopatías se hace mediante exploración física si son accesibles, o mediante TAC o RM si no lo son. En el momento de la cirugía deben biopsiarse los ganglios ipsilaterales y en los tumores abdominales también los contralaterales. Todo ganglio mayor de 2 cm en tórax o abdomen debe biopsiarse y, si no se puede, se asume que es positivo.
Estadios
• Estadio 1: tumor localizado y quirúrgicamente resecable. Se permite la existencia de enfermedad microscópica residual. Los ganglios pegados o extirpados junto al tumor pueden ser positivos.
• Estadio 2 A: tumor localizado con resección quirúrgica incompleta. Ganglios linfáticos ipsilaterales no adheridos al tumor negativos.
• Estadio 2 B: tumor localizado con resección quirúrgica completa o incompleta, con ganglios ipsilaterales no adheridos al tumor positivos. Los contralaterales son negativos.
• Estadio 3: tumor primario unilateral que infiltra a través de la línea media, con o sin adenopatías regionales positivas. Tumor unilateral localizado con adenopatías positivas contralaterales. Tumor de la línea media con extensión bilateral por infiltración (irresecable) o con adenopatías positivas.
• Estadio 4: metástasis en ganglios linfáticos a distancia, huesos, médula ósea, hígado, piel y otros órganos.
• Estadio 4 S: tumor primario localizado (definido según estadio 1, 2 A, 2B) con diseminación limitada a piel, hígado y médula ósea (siempre menor al 10% de las células nucleadas y con MIBG negativo de médula ósea) (2,3,59).
Existen evidencias sobre los pacientes incluidos en estadio 4 por afectación ganglionar a distancia, del hígado o de médula ósea (excluyendo los incluidos en el 4S), especialmente si son menores de dos años, tienen mejor pronóstico que los incluidos por afectación ósea. Debido a que esta distinción puede influir en el pronóstico o en la elección del tratamiento, los criterios por los que los pacientes se incluyen en estadio 4 deben ser registrados. Sin embargo, estas diferencias pueden verse afectadas o desaparecer por avances en el tratamiento (60). El estadio 4S ha seguido considerándose como diferente, basándose en los resultados favorables que generalmente se consiguen con estos pacientes, y por las evidencias recientes que los diferencian en cuanto a la biología molecular (61).
Consideraciones pronósticas
Factores clínicos: la edad, el estadio y la localización del tumor primario son los factores pronóstico más importantes. La supervivencia libre de enfermedad (SLE) es de un 80-90% en los estadios 1, 2 y 4S. En los estadios 3 y 4, el pronóstico depende de la edad, los lactantes con tumores estadio 3 tienen una SLE a los 3 años del 80%, mientras que en niños mayores es < 50%. En el estadio 4, la SLE es del 50-60% en menores de un año y no llega al 15% en los mayores (5,22,62).
Factores bioquímicos: un aumento de la LDH indica un crecimiento tumoral rápido o turn-over celular acelerado y, aunque el dato es muy inespecífico, se sabe que una LDH > 1.500 mg/ml se asocia a un mal pronóstico. Un índice de HVA/VMA en orina de 24 horas > 1,5 se asocia a un mal pronóstico, así como la ausencia de catecolaminas en orina.
Los dos factores reflejan que el tumor es muy indiferenciado. Una ferritina > 250 ng/ml se asocia a tumores con crecimiento rápido o a tumores grandes. Parece ser que la célula tumoral necesita hierro o ferritina para su metabolismo.
La enolasa neuronal específica (NSE) es una proteína intracitoplasmática que no es específica de tumores neuronales, pero tiene valor como marcador tumoral en el seguimiento de la enfermedad y detección de recidivas. Sólo tiene valor pronóstico si es > de 100 ng/ml. Los gangliósidos son gliceroesfingolípidos que contienen ácido siálico se detectan no sólo en las células tumorales, sino también en el plasma de los enfermos y, por tanto, pueden ser útiles para detectar masa tumoral mínima. El aumento de niveles de GD2 en suero asocia mal pronóstico (63).
Factores histológicos: la Clasificación Internacional Patológica del Neuroblastoma (INPC), basada en la clasificación de Shimada, tiene valor pronóstico. Asimismo, si en el estudio de inmunohistoquímica se observa un aumento de CD44, el pronóstico es favorable (tumor diferenciado).
Factores genéticos: existen una serie de datos moleculares (N-MYC amplificado, LOH 1p, índice de ADN o DI < 1), cuya expresión se asocia a un peor pronóstico y que caracterizan a un tumor biológicamente más agresivo y refractario al tratamiento. La amplificación del N-MYC se observa en tumores avanzados, se asocia a progresión tumoral y es un dato de mal pronóstico, independientemente de la edad del niño o del estadio del tumor. El hallazgo de una deleción del 1p también confiere un mal pronóstico y, en general, se asocia a una amplificación del N-MYC. Más recientemente, se ha observado que la expresión de m-TRK A indica diferenciación neuronal y, por tanto, se asocia a un mejor pronóstico. Otros genes estudiados, como el MDR1, MRP, nm23 y CD44 tienen resultados variables (64,65). Por último, la hiperdiploidía o DI (índice de ADN) alto se asocia a un buen pronóstico y es característica de lactantes sin otras alteraciones genéticas en el tumor (66,67).
En resumen, desde el punto de vista genético, el neuroblastoma es una enfermedad heterogénea. Se ha propuesto la siguiente clasificación:
1. Tumores hiperdiploides o triploides, sin amplificación del N-MYC, ni de leción del 1p y con una expresión alta de Trk A. Estos tumores suelen presentarse en el lactante, localizados (1 y 2) o en estadio 4S y tienen un pronóstico favorable (> 90% se curan). Estos son los tumores que suelen detectarse en los programas de screening.
2. Tumores casi diploides o tetraploides, sin amplificación del N-MYC pero con alguna anomalía estructural y en los que no hay expresión del Trk A o es muy baja. Estos tumores se suelen observar en pacientes mayores de un año, en estadios 3 y 4 y tienen un pronóstico intermedio, con una supervivencia global del 25-50%.
3. Tumores con amplificación del N-MYC, en general casi diploides o tetraploides con 1p LOH. La expresión de Trk A es baja o ausente. Los enfermos son mayores de un año y los tumores en estadios 3 y 4. En general, responden al tratamiento sólo transitoriamente y su pronóstico es malo (< 5% de supervivencia a los dos años) (1,69,70).
Tamizaje de neuroblastoma
El neuroblastoma del lactante tiene mucho mejor pronóstico que el del niño mayor de un año, por tanto, si se diagnostica este tumor antes del año de edad, podrían identificarse niños potencialmente más curables. Con este objetivo, se inició en Japón en los años 80, un cribaje poblacional en lactantes de 3 y 6 meses de edad. Posteriormente, se realizaron estudios en Europa, EE.UU. y Canadá con el fin de comprobar la factibilidad y utilidad del cribaje poblacional (71,72). En 1988, el grupo de estudio de cribaje del neuroblastoma llegó a la conclusión de que debía suspenderse por los siguientes motivos:
• Se desconocen datos sobre la historia natural del neuroblastoma.
• El tamaño del grupo poblacional (si es que lo hay) que se beneficiaría de dicho cribaje es, de momento, desconocido.
• El tiempo de evolución del neuroblastoma es desconocido.
• Existe un riesgo de diagnósticos en exceso, que conlleva tratamientos innecesarios y potencialmente tóxicos.
• El cribaje del neuroblastoma se sigue de un impacto psicosocial importante en las familias de los lactantes diagnosticados.
• No hay evidencia de una reducción en la mortalidad del neuroblastoma imputable al cribaje.
Análisis y Conclusión
Los neuroblastomas son tumores neuroblásticos de los niños, con una edad media de diagnóstico de unos 15 a 17 meses. Los factores causales no están bien definidos, aunque diversos trastornos genéticos ocasionalmente se asocian con el desarrollo de los neuroblastomas (2,72-74). Son tumores clínicamente heterogéneos, variando en localización, aspecto histopatológico y las características biológicas. Los neuroblastomas son notables por su evolución natural altamente variable, que puede ir desde una regresión espontánea, a la maduración de una ganglioneuroma benignos o enfermedad agresiva con diseminación metastásica que conduce a la muerte (28,74-76).
Las mejoras en los resultados en niños con neuroblastoma ha sido el resultado del grupo cooperativo, los ensayos clínicos multicéntricos, que han integrado enfoques de modalidad combinada con una comprensión de los factores pronósticos que afectan el resultado (51,77,78). Los pacientes deben ser manejados en un ambiente donde la experiencia adecuada en el tratamiento del neuroblastoma se encuentra disponible.
La patogénesis molecular de los neuroblastomas se ha estudiado de forma exhaustiva y la información acerca de las anormalidades específicas es un componente importante de la definición de grupos de riesgo pronósticos (79-81):
Las deleciones cromosómicas, en particular del cromosoma 1p, están asociados con un mal pronóstico, con la amplificación del oncogén MYCN.La presencia o ausencia de anormalidades cromosómicas y la amplificación de MYCN se han incorporado al analisis y estratificación por riesgo para neuroblastomas, lo cual es importante para determinar el tratamiento adecuado para los pacientes recién diagnosticados (31,39,82-84).
Los principales factores que influyen en el comportamiento clínico de los neuroblastomas son el estadio tumoral, la edad al momento del diagnóstico, la clasificación de riesgo patológico, citogenética y genética molecular. Estos factores se han combinado para definir los grupos de bajo, intermedio y alto riesgo, que se utilizan para definir las estrategias de tratamiento (54,85-89).
Para los niños con enfermedad de bajo riesgo, la cirugía es el tratamiento primario cuando la resección completa es posible. Para los pacientes con tumores de bajo riesgo que no se pueden resecar completamente o que tienen complicaciones potencialmente mortales, la quimioterapia y / o terapia de radiación puede ser requerida (16,84,90-93).
En el subgrupo de pacientes con enfermedad en estadio 4S, la observación puede ser una opción, puesto que hay una alta tasa de regresión espontánea. Para los niños con enfermedad de riesgo intermedio, un enfoque de modalidad combinada que incluye la resección quirúrgica y quimioterapia es estándar (94-98). El papel de la radioterapia es menos claro, excepto en el contexto de la evolución de la enfermedad a pesar de la quimioterapia más cirugía o por complicaciones como la compresión de la médula espinal. Para los niños con neuroblastoma de alto riesgo, las mejoras sustanciales en el largo plazo los resultados se han visto con un enfoque mas agresivo mediante una modalidad combinada (67,69,98-100). Estos enfoques generalmente incluyen quimioterapia, resección quirúrgica, quimioterapia de dosis alta con rescate de células madre y terapia de radiación. Esta forma de tratamiento ha mejorado los resultados a largo plazo. Los pacientes pediatricos que han recibido tratamiento para el neuroblastoma corren el riesgo de recurrencia y de complicaciones tardías de su tratamiento (17,101,102). El tratamiento medico debe ser biopsicosocial con un seguimiento multidisciplinario, evaluando los posibles problemas a largo plazo y mediano plazo.
Referencias
1. Carrasquillo JA, Pandit-Taskar N, Chen CC. Radionuclide therapy of adrenal tumors. J Surg Oncol. 2012 Jun; 20. [ Links ]
2. Asgharzadeh S, Pique-Regi R, Sposto R, et al. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J Natl Cancer Inst. 2006; 98: 1193-203. [ Links ]
3. Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003; 3: 895-902. [ Links ]
4. Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006; 6: 425-36. [ Links ]
5. Bown N. Neuroblastoma tumour genetics: Clinical and biological aspects. JClin Pathol. 2001; 54: 897-910. [ Links ]
6. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nature Reviews/cancer. 2003; 3: 203-16. [ Links ]
7. Brodeur GM, Maris JM. Neuroblastoma. En: Pizzo PA, Poplack DG, Eds. Principles and Practice of Pediatric Oncology. 4ª ed. Philadelphia: Lippincott Williams & Wilkins; 2001. [ Links ]
8. López-Ibor B, Moreno L. Tumores de cresta neural. Hematología y oncología ediátricas. Madero López L, Muñoz Villa A. Madrid: Ergon; 2006. [ Links ]
9. Simon T, Hero B, Benz-Bohm G, Von Schweinitz D, Berthold F. Review of image defined risk factors in localized neuroblastoma patients. Results of the GPOH NB97 Trial. Pediatr Blood Câncer 2008; 50 (5): 965-9. [ Links ]
10. Paramasivam A, Sambantham S, Shabnam J, et al. Anti-cancer effects of thymoquinone in mouse neuroblastoma (Neuro-2a) cells through caspase-3 activation with down-regulation of XIAP. Toxicol Lett. 2012 Jun 22. [ Links ]
11. Lee MH, Yoo SY, Kim JH, et al. Hypervascular hepatic nodules in childhood cancer survivors: clinical and imaging features. Clin Imaging. 2012 Jul; 36(4): 301-7. [ Links ]
12. Zwaveling S, Tytgat GA, van der Zee DC, et al. Is complete surgical resection of stage 4 neuroblastoma a prerequisite for optimal survival or may >95 % tumour resection suffice? Pediatr Surg Int. 2012 Jun 22. [ Links ]
13. Grosfeld JL. Risk-based management of solid tumors in children. Am J Surg. 2000 Nov; 180: 322-7. [ Links ]
14. Berthold F, Hero B. Neuroblastoma: current drug therapy recommendations as part of the total treatment approach. Drugs. 2000 Jun; 59 (6): 1261-77. [ Links ]
15. Cheung NV, Kushner BH, Kramer K. Monoclonal antibodybased therapy of neuroblastoma. Hematol Oncol Clin North Am. 2001 Oct; 15 (5): 853-66. [ Links ]
16. Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. N Engl J Med. 1999 Oct 14; 341 (16): 1165-73. [ Links ]
17. Hoefnagel CA. Nuclear Medicine Therapy of Neuroblastoma. Q J Nucl Med. 1999 Dec; 43: 336-43. [ Links ]
18. Evans AE, D'Angio GJ, Sather HN, de Lorimer AA, Dalton A, Ungerleider RS, et al. A comparison of four staging systems for localized and regional neuroblastoma: a report from the children's cancer study group. J Clin Onc. 1990 Apr; 8 (4): 678-688. [ Links ]
19. Partrick D, Bensar D, Geiger, JD, Teitelbaum, DH. Successful throacoscopic lung biopsy in children utilizing preoperative CT-guided localization. J Pediatr Surg. 2002; 37(7): 970-973. [ Links ]
20. Geiger JD, Hutchinson RJ, Hohenkirk LF, McKenna EA, Yanik GA, Levine JE, et al. Vvaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific t cells and mediate tumor regression. Cancer Research. 2001; 61, 8513-8519. [ Links ]
21. Hemmila MR, Foley DS, Castle VP, Hirschl RB. The response to splenectomy in pediatric patients with ideopathic thrombocytopenic purpura who fail high-dose intravenous immune globulin. J Ped Surg. 2000; 35(6): 967-72. [ Links ]
22. Azizkhan RG, Rescorla FJ, Haase GM, Applebaum H, Dillon PW, Coran AG, et al. Diagnosis, management and outcome of teratomas in neonates and infants: a multi-institutional study. Paediatr Croat. 1999; 43: 163-171. [ Links ]
23. Barnhart DC, Hirschl RB, Garver KA, Geiger JD, Harmon CM, Coran AG. Conservative management of mesenchymal hamartoma of the liver. J Ped Surg. 1997; 32:1495-1498. [ Links ]
24. Geiger JD. Surgery for hepatoblastoma in children, current opinion in pediatrics, 1996; 8(3)282-290. [ Links ]
25. Dillion P, Whalen T, Azizkhan R, Haase G, Coran AG, King D, Smith M. Neonatal soft tissue sarcomas: The influence of pathology on treatment and survival. J Ped Surg. 1995; 30: 1038-1041. [ Links ]
26. Geiger JD, Hutchinson RJ, Hohenkirk LF, McKenna E, Chang A, Mul, J. Treatment of solid tumours in children with tumour lysate-pulsed dendritic cells. The Lancet. 2000; 356(9236): 1163-1164. [ Links ]
27. Schmeling DJ, Coran AG. Wilms' Tumor (Nephroblastoma). Pediatric Rounds 1988; 8:3-5. [ Links ]
28. Evans AE, D'Angio GJ, Randolph J. A proposed staging for children with neuroblastoma: Children's Cancer Study Group A. Cancer. 1971; 27: 374-378. [ Links ]
29. Wilson LM, Draper GJ. Neuroblastoma, its natural history and prognosis: A study of 487 cases. BMJ. 1974; 3: 301-307. [ Links ]
30. Shimada H, Chatten J, Newton WA Jr, et al. Histopathologic prognostic factors in neuroblastic tumors: Definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J Natl Cancer Inst. 1984; 73: 405-416. [ Links ]
31. Brodeur GM, Seeger RC, Schwab M, et al. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984; 224: 1121-1124. [ Links ]
32. Seeger RC, Brodeur GM, Sather H, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med.1985; 313: 1111-1116. [ Links ]
33. Kushner BH, Cheung NK, LaQuaglia MP, et al. International neuroblastoma staging system stage 1 neuroblastoma: A prospective study and literature review. J Clin Oncol. 1996; 14: 2174-2180. [ Links ]
34. Nitschke R, Smith EI, Shochat S, et al. Localized neuroblastoma treated by surgery: A Pediatric Oncology Group study. J Clin Oncol. 1988; 6: 1271-1279. [ Links ]
35. Alvarado CS, London WB, Look AT, et al. Natural history and biology of stage A neuroblastoma: A Pediatric Oncology Group study. J Pediatr Hematol Oncol. 2000; 22: 197-205. [ Links ]
36. Perez CA, Matthay KK, Atkinson JB, et al. Biologic variables in the outcome of stages I and II neuroblastoma treated with surgery as primary therapy: A Children's Cancer Group study. J Clin Oncol. 2000; 18: 18-26. [ Links ]
37. Matthay KK, Villablanca JG, Seeger RC, et al. Treatment of highrisk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid: Children's Cancer Group. N Engl J Med. 1999; 341: 1165-1173. [ Links ]
38. Bowman LC, Castleberry RP, Cantor A, et al. Genetic staging of unresectable or metastatic neuroblastoma in infants: A Pediatric Oncology Group study. J Natl Cancer Inst. 1997; 89: 373-380. [ Links ]
39. Schmidt ML, Lukens JN, Seeger RC, et al. Biologic factors determine prognosis in infants with stage IV neuroblastoma: A prospective Children's Cancer Group study. J Clin Oncol. 2000; 18: 1260-1268. [ Links ]
40. Schwab M, Alitalo K, Klempnauer KH, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983; 305:245-248. [ Links ]
41. Shapiro DN, Valentine MB, Rowe ST, et al. Detection of N-myc gene amplification by fluorescence in situ hybridization: Diagnostic utility for neuroblastoma. American J Pathol. 1993; 142: 1339-1346. [ Links ]
42. Look AT, Hayes FA, Nitschke R, et al. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N Engl J Med. 1984; 311: 231-235. [ Links ]
43. Brodeur GM, Pritchard J, Berthold F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993; 11: 1466-1477. [ Links ]
44. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958; 53: 457-481. [ Links ]
45. Matthay KK, Perez C, Seeger RC, et al. Successful treatment of stage III neuroblastoma based on prospective biologic staging: A Children's Cancer Group study. J Clin Oncol. 1998: 16: 1256-1264. [ Links ]
46. Kushner BH, Cheung NK, LaQuaglia MP, et al. Survival from locally invasive or widespread neuroblastoma without cytotoxic therapy. J Clin Oncol. 1996; 14: 373-381. [ Links ]
47. Cheung NK, Kushner BH, LaQuaglia MP, et al. Survival from non-stage 4 neuroblastoma without cytotoxic therapy: An analysis of clinical and biological markers. Eur J Cancer. 1997; 33: 2117-2120. [ Links ]
48. Kushner BH, Kramer K, LaQuaglia MP, et al. Curability of recurrent disseminated disease after surgery alone for local-regional neuroblastoma using intensive chemotherapy and anti-G(D2) immunotherapy. J Pediatr Hematol Oncol. 2003; 25: 515-519. [ Links ]
49. Evans AE, Silber JH, Shpilsky A, et al. Successful management of low-stage neuroblastoma without adjuvant therapies: A comparison of two decades, 1972 through 1981 and 1982 through 1992, in a single institution. J Clin Oncol. 1996; 14: 2504-2510. [ Links ]
50. Matthay KK, Sather HN, Seeger RC, et al. Excellent outcome of stage II neuroblastoma is independent of residual disease and radiation therapy. J Clin Oncol. 1989; 7: 236-244. [ Links ]
51. De Bernardi B, Conte M, Mancini A, et al. Localized respectable neuroblastoma: Results of the second study of the Italian Cooperative Group for Neuroblastoma. J Clin Oncol. 1995; 13: 884-893. [ Links ]
52. Castleberry RP, Cantor AB, Green AA, et al. Phase II investigational window using carboplatin, iproplatin, ifosfamide, and epirubicin in children with untreated disseminated neuroblastoma: A Pediatric Oncology Group study. J Clin Oncol. 1994; 12: 1616-1620. [ Links ]
53. Pinkerton CR, Zucker JM, Hartmann O, et al. Short duration, high dose, alternating chemotherapy in metastatic neuroblastoma: ENSG 3C induction regimen -The European Neuroblastoma Study Group. Br J Cancer.1990; 62: 319-323. [ Links ]
54. Campbell LA, Seeger RC, Harris RE, et al. Escalating dose of continuous infusion combination chemotherapy for refractory neuroblastoma. J Clin Oncol. 1993; 11: 623-629. [ Links ]
55. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009; 41: 1238-42. [ Links ]
56. Gangemi RM,Griffero F, Marubbi D, Perera M,Capra MC, Malatesta P, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009; 27: 40-8. [ Links ]
57. Rodriguez-Pinilla SM, Sarrio D, Moreno-Bueno G, Rodriguezil Y, Martinez MA,Hernandez L, et al. Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Mod Pathol. 2007; 20: 474-81. [ Links ]
58. Phi JH, Park SH, Kim SK, Paek SH, Kim JH, Lee YJ, et al. Sox2 expression in brain tumors: a reflection of the neuroglial differentiation pathway. Am J Surg Pathol. 2008; 32: 103-12. [ Links ]
59. Riggi N, Suva ML, De Vito C, Provero P, Stehle JC, Baumer K, et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev 2010; 24: 916-32. [ Links ]
60. Kim Y, Lin Q, Zelterman D, Yun Z . Hypoxia-regulated delta-like 1 homologue enhances cancer cell stemness and tumorigenicity. Cancer Res. 2009; 69: 9271-80. [ Links ]
61. Melone MA, Giuliano M, Squillaro T, Alessio N, Casale F, Mattioli E, et al. Genes involved in regulation of stem cell properties: studies on their expression in a small cohort of neuroblastoma patients. Cancer Biol Ther. 2009; 8: 1300-6. [ Links ]
62. Carlsson P, Mahlapuu M . Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002; 250: 1-23. [ Links ]
63. Kalin TV, Wang IC, Ackerson TJ, Major ML, Detrisac CJ, Kalinichenko VV, et al. Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006; 66: 1712-20. [ Links ]
64. Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF, et al. Fox- M1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006; 66: 3593-602. [ Links ]
65. Major ML, Lepe R, Costa RH. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation- dependent recruitment of p300/CBP coactivators. Mol Cell Biol. 2004; 24: 2649-61. [ Links ]
66. Albino D, Scaruffi P, Moretti S, Coco S, Truini M, Di Cristofano C, et al. Identification of low intratumoral gene expression heterogeneity in neuroblastic tumors by genome-wide expression analysis and game theory. Cancer. 2008; 113: 1412-22. [ Links ]
67. De Preter K, Vandesompele J, Heimann P, et al. Human fetal neuroblast and neuroblastoma transcriptome analysis confirms neuroblast origin and highlights neuroblastoma candidate genes. Genome Biol. 2006; 7: R84. [ Links ]
68. Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006; 313: 1929-35. [ Links ]
69. Wang Q, Diskin S, Rappaport E, et al. Integrative genomics identifies distinct molecular classes of neuroblastoma and shows that multiple genes are targeted by regional alterations in DNA copy number. Cancer Res. 2006; 66: 6050-62. [ Links ]
70. McArdle L, McDermott M, Purcell R, et al. Oligonucleotide microarray analysis of gene expression in neuroblastoma displaying loss of chromosome 11q. Carcinogenesis. 2004; 25: 1599-609. [ Links ]
71. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5: R80. [ Links ]
72. Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001; 98: 5116-21. [ Links ]
73. Breitling R, Armengaud P, Amtmann A, Herzyk P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004; 573: 83-92. [ Links ]
74. De Preter K, Barriot R, Speleman F, Vandesompele J, Moreau Y. Positional gene enrichment analysis of gene sets for high-resolution identification of overrepresented chromosomal regions. Nucleic Acids Res. 2008; 36: e43. [ Links ]
75. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102: 15545-50. [ Links ]
76. Muhlethaler-Mottet A, Meier R, Flahaut M, et al. Complex molecular mechanisms cooperate to mediate histone deacetylase inhibitors anti-tumour activity in neuroblastoma cells. Mol Cancer. 2008; 7: 55. [ Links ]
77. Johnsen JI, Segerstrom L, Orrego A, et al. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growthin vitro and in vivo. Oncogene. 2008; 27: 2910-22. [ Links ]
78. Al Shaer L, Walsby E, Gilkes A, et al. Heat shock protein 90 inhibition is cytotoxic to primary AML cells expressing mutant FLT3 and results in altered downstream signalling. Br J Haematol. 2008; 141: 483-93. [ Links ]
79. Ryhanen T, Mannermaa E, Oksala N, et al. Radicicol but not geldanamycin evokes oxidative stress response and efflux protein inhibition in ARPE-19 human retinal pigment epithelial cells. Eur J Pharmacol. 2008; 584: 229-36. [ Links ]
80. Lefever S, Vandesompele J, Speleman F, Pattyn F. RTPrimerDB: the portal for real-time PCR primers and probes. Nucleic Acids Res. 2009; 37: D942-5. [ Links ]
81. Pattyn F, Robbrecht P, De Paepe A, Speleman F, Vandesompele J. RTPrimerDB: the real-time PCR primer and probe database, major update 2006. Nucleic Acids Res. 2006; 34: D684-8. [ Links ]
82. Pattyn F, Speleman F, De Paepe A, Vandesompele J. RTPrimerDB: the real-time PCR primer and probe database. Nucleic Acids Res. 2003; 31: 122-3. [ Links ]
83. Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007; 8: R19. [ Links ]
84. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002; 3: RESEARCH0034. [ Links ]
85. Mosse YP, Diskin SJ, Wasserman N, et al. Neuroblastomas have distinct genomic DNA profiles that predict clinical phenotype and regional gene expression.Genes Chromosomes Cancer. 2007; 46: 936-49. [ Links ]
86. Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002; 99: 6567-72. [ Links ]
87. Janoueix-Lerosey I, Novikov E, Monteiro M, et al. Gene expression profiling of 1p35-36 genes in neuroblastoma. Oncogene. 2004; 23: 5912-22. [ Links ]
88. Lastowska M, Viprey V, Santibanez-Koref M, et al. Identification of candidate genes involved in neuroblastoma progression by combining genomic and expression microarrays with survival data. Oncogene. 2007; 26: 7432-44. [ Links ]
89. Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005; 352: 254-66. [ Links ]
90. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006; 6: 38-51. [ Links ]
91. Tang XX, Robinson ME, Riceberg JS, et al. Favorable neuroblastoma genes and molecular therapeutics of neuroblastoma. Clin Cancer Res. 2004; 10: 5837-44. [ Links ]
92. Hahn CK, Ross KN, Warrington IM, et al. Expression-based screening identifies the combination of histone deacetylase inhibitors and retinoids for neuroblastoma differentiation. Proc Natl Acad Sci U S A. 2008; 105: 9751-6. [ Links ]
93. Kang J, Kamal A, Burrows FJ, Evers BM, Chung DH. Inhibition of neuroblastoma xenograft growth by Hsp90 inhibitors. Anticancer Res. 2006; 26: 1903-8. [ Links ]
94. Bagatell R, Beliakoff J, David CL, Marron MT, Whitesell L. Hsp90 inhibitors deplete key anti-apoptotic proteins in pediatric solid tumor cells and demonstrate synergistic anticancer activity with cisplatin. Int J Cancer. 2005; 113: 179-88. [ Links ]
95. Bagatell R, Gore L, Egorin MJ, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-N-allylamino-17-demethoxygeldanamycin in pediatric patients with recurrent or refractory solid tumors: a pediatric oncology experimental therapeutics investigators consortium study. Clin Cancer Res. 2007; 13: 1783-8. [ Links ]
96. Sharma SV, Agatsuma T, Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene. 1998; 16: 2639-45. [ Links ]
97. Misawa A, Hosoi H, Tsuchiya K, Sugimoto T. Rapamycin inhibits proliferation of human neuroblastoma cells without suppression of MycN. Int J Cancer. 2003; 104: 233-7. [ Links ]
98. Gil-Ad I, Shtaif B, Levkovitz Y, Dayag M, Zeldich E, Weizman A. Characterization of phenothiazine-induced apoptosis in neuroblastoma and glioma cell lines: clinical relevance and possible application for brain-derived tumors. J Mol Neurosci. 2004; 22: 189-98. [ Links ]
99. Yang J, Shamji A, Matchacheep S, Schreiber SL. Identification of a small-molecule inhibitor of class Ia PI3Ks with cell-based screening. Chem Biol. 2007; 14: 371-7. [ Links ]
100. Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008; 1784: 159-85. [ Links ]
