










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Med
versão impressa ISSN 0121-5256
rev.fac.med vol.25 no.1 Bogotá jan./jun. 2017
https://doi.org/10.18359/rmed.2925
ARTÍCULO DE REVISIÓN
Doi: http://dx.doi.org/10.18359/rmed.2925
COMPROMISO PULMONAR EN LA ENFERMEDAD HEPÁTICA: UNA ACTUALIZACIÓN
PULMONARY COMMITMENT IN HEPATIC DISEASE
COMPROMISSO PULMONAR NA DOENÇA HEPÁTICA: UMA ATUALIZAÇÃO
JUAN FELIPE VASQUEZ RODRIGUEZ MD1*, JAMES MILTON YURGAKI MD2, ROBIN RADA ESCOBAR MD3
1Residente de tercer año de Medicina Interna, Universidad Militar Nueva Granada
Hospital Militar Central - Bogotá, Colombia
2Médico Internista, Endocrinólogo y Gastroenterologo
Hospital Militar Central - Bogotá, Colombia
3Médico internista Neumologo, jefe del Servicio de Medicina Interna
Hospital Militar Central - Bogotá, Colombia
*Correspondencia: juanfevasquez8@gmail.com
Recibido: 4 Junio 2016 Aceptado: 13 Febrero 2017
RESUMEN
La enfermedad Hepática es una causa importante de morbilidad y Mortalidad en el mundo; asociada a compromiso orgánico múltiple. En el pulmón, tres condiciones clínicas han sido descritas; estos cuadros Incluyen, el Síndrome Hepatopulmonar (SHP), que obedece a un trastorno de la oxigenación por dilatación de la vasculatura pulmonar. La hipertensión portopulmonar (HPP), mediada por desequilibrio entre agentes vasodilatadores y vasoconstrictores que conducen a un aumento de la presión media de la arteria pulmonar y por último el Hidrotórax Hepático (HH), que es la condición menos prevalente, se fundamenta principalmente en anomalías anatómicas del diafragma, con o sin relación a la presencia de ascitis. La presencia SHP o HPP es predictor independiente de mortalidad, resaltando su importancia en la elegibilidad de pacientes para trasplante hepático ortotópico como medida curativa.
Palabras Clave: Cirrosis, Síndrome Hepatopulmonar, Hipertensión Portopulmonar, Hidrotórax Hepático, Trasplante HepáticoABSTRACT
Liver disease is a major cause of morbidity and mortality in the world; it is associated with multiple organ involvement. In the lung, three clinical conditions are described; these conditions include, Hepatopulmonary Syndrome (HPS), which is due to a oxygenation defect by the development of pulmonary vascular dilatation. Portopulmonary (HPP) hypertension, it's mediated by an imbalance between vasodilator and vasoconstrictor agents leading to an increase in mean pulmonary artery pressure and finally the Hydrothorax Liver (HH), that is the least prevalent condition, it's based on anatomical diaphragm abnormalities, with or without ascites. The presence of SHP or HPP is an independent predictor of mortality, highlighting its importance in the eligibility of patients for orthotopic liver transplantation as a curative measure.
Keywords: Cirrhosis, Hepatopulmonary syndrome, hypertension portopulmonary, Hydrothorax Liver, Liver TransplantationRESUMO
A doença hepatica é uma das principais causas de morbidade e mortalidade no mundo; está associado ao compromisso de vários órgãos. No pulmão, três condições clínicas são descritas; estas condições incluem, Síndrome Hepatopulmonar (HPS), que é devido a um defeito de oxigenação do dilatação vascular pulmonar. A hipertensão portopulmonar (HPP), é mediada por um desequilíbrio entre vasodilatadores e agentes vasoconstritores, levando a um aumento da pressão arterial média da artéria pulmonar e, finalmente, ao hidrotorax hepatico (HH), essa é a condição menos prevalente, é baseada em anormalidades anatômicas do diafragma, com ou sem ascite. A presença de SHP ou HPP é um preditor independente de mortalidade, destacando sua importância na elegibilidade de pacientes para transplante de hepatico como medida curativa.
Palavras-chave: Cirrose; Síndrome hepatopulmonar; Hipertensão portopulmonar; Hidrotorax Hepatico; Transplante de fígado.
Introducción
El Hígado es uno de los principales órganos en la homeostasis sistémica y su disfunción puede conducir a compromiso multiorgánico, con consecuencias devastadoras. La cirrosis, es una las principales condiciones relacionadas al compromiso Hepático y guarda clara asociación con el desarrollo de dificultad respiratoria e hipoxia; los pacientes con esta enfermedad frecuentemente se presentan con acropaquia, taquipnea e hiperventilación; estas manifestaciones se correlacionan con la presencia de alteraciones de la oxigenación, desequilibrio ventilación/perfusión, compromiso en Pruebas de función pulmonar y desarrollo de Efusiones Pleurales (1).
La Cirrosis, constituye una causa importatnte de Morbi-mortalidad a nivel mundial, se estima que es la décimo cuarta causa de muerte en el mundo, (2) con estimados que van desde 1% hasta el 57% a un año, según su estadio (3); aportando para el año 2010 el 2% del total de fallecimientos que corresponde a 1.030.800 muertos (4). En Estados Unidos su prevalencia es del 0.27% (633.323 adultos), de los cuales el 69% no es consciente de su condición (5). Esta Enfermedad le cuesta al estado americano un monto total anual oscilante entre los 14 millones y los 2 billones de dólares (6).
En los países desarrollados la etiología más frecuente de cirrosis es la infección crónica por virus de hepatitis C y B; el Consumo del alcohol es la principal causa de esta enfermedad en los países en vía de desarrollo (6).
Para la población Colombiana, el Comportamiento epidemiológico de la enfermedad hepática esta pobremente establecido, un estudio de Montoya et al, publicado en el 2012, realizó una caracterización del comportamiento de estas enfermedades en el la región Centro - Occidental del territorio nacional, encontrando como principal causa de cirrosis la ingesta de alcohol (24%), seguido de hepatitis autoinmune (14%), cirrosis criptogénica (12%), esteatohepatitis no alcohólica NASH (11%), cirrosis biliar primaria (9%), infección por virus de la hepatitis B (8%), infección por virus de la hepatitis C (7%), hepatocarcinoma (6%), y otras trastornos como enfermedad de Wilson, hemocromatosis, Cirrosis por cardiopatía y degeneración Carvenomatosa de la porta que generan 6% adicional (7).
El compromiso de la función pulmonar en la hepatopatía crónica es multifactorial, incluye la descompensación de enfermedades pre existentes como EPOC, cardiopatías y enfermedad pulmonar intersticial, así como condiciones secundarias a la enfermedad hepática, tales como: Síndrome Hepatopulmonar (SHP), Hipertensión Portopulmonar (HPP) e Hidrotórax Hepático (HH) (8).
Las manifestaciones pulmonares de las enfermedades hepáticas autoinmunes son independientes del trastorno de la función hepática. Por último, los pacientes con cirrosis tienen mayor predisposición a neumonía bacteriana; además presentan restricción de la mecánica ventilatoria por la excursión limitada del diafragma secundaria a la presencia de ascitis (9).
Síndrome Hepatopulmonar (SHP)
La primera descripción de este Síndrome data de 1884, tras el estudio de Flückiger de una Mujer con cirrosis, cianosis y acropaquia. Posteriormente Ryedell en 1956 y Berthelot, en 1966 continuaron las observaciones de alteraciones a nivel vascular pulmonar en autopsias de pacientes con cirrosis (10).
El Término empleado por primera vez por Kennedy y Knudson en 1977 y hace referencia a un defecto de Oxigenación en presencia de Enfermedad Hepática, el cual es causado por dilatación Vascular Intrapulmonar; éste síndrome consta de una triada: alteración en la Oxigenación, Enfermedad Hepática y Dilatación de vasculatura Intrapulmonar (11,12).
Su prevalencia oscila entre 10% a 30%, siendo la mayoría de los casos asintomático, razón por la cual puede llegar a pasarse por alto (13), no hay relación entre la severidad del SHP y la enfermedad hepática evaluada por la clasificación de Child-Turcotte-Pugh, o por el Modelo para la Enfermedad Hepática en Estadio Terminal (Model for End-Stage Liver Disease - MELD) (14).
La presencia de SHP en el paciente cirrótico es un factor independiente de mortalidad; el pronóstico de la cirrosis es mucho peor en aquellos que cursan con SHP, según el estudio realizado por Schenk P, et al, (15), se determinó que la sobrevida media de los pacientes con SHP comparado con aquellos sin la enfermedad fue de 10.6 meses y 40.8 meses respectivamente (p< 0.05). Adicionalmente, el estudio permitió establecer que el SHP confiere peor pronóstico para todos los estadios de Child - Pugh, así para los pacientes CHILD C, la sobrevida media fue de 14,7 meses sin SHP VS 2.9 meses en pacientes con SHP (P < 0.05), con igual tendencia en los pacientes con CHILD B, pero sin significancia estadística.
La mortalidad es más elevada al alcanzar valores de PaO2 menores de 50 mmHg, aun así, la muerte por hipoxemia severa o falla ventilatoria es rara y cuando ocurre, se asocia otras complicaciones de la enfermedad hepática (16).
Fisiopatología
El desarrollo de la Dilatación de la Vasculatura Pulmonar tiene como principal factor un aumento considerable en las concentraciones de Óxido Nítrico (principal factor), y otros agentes vasoactivos.
El modelo fisiopatológico del SHP se basa en tres postulados: (16-18).
- Daño Hepático asociado al desarrollo de Endotoxemia y sobreproducción de Agentes Vasodilatadores (Evidencia de sobreexpresión Óxio Nítrico Sintasa Inducible (iNOS) y Óxido NItrico SIntasa Endotelias (eNOS), en relación a Incremento en los Niveles de Lipopolisacárido (lps): Endotoxemia Intestinal)
- Producción Hepática de vasodilatadores: Prostacilcina, Prostaglandina E1, Prostaglandina E2, Óxido Nítrico (ON), Péptido Intestinal Vasoactivo, Calcitonina Y glucagón.
- Incremento en la Expresión eNOS, así como de iNOS, las cuales obedecen a un aumento en la Producción Hepática de Endotelina 1, y Factor de Necrosis tumoral (TNFa)
El incremento en la Endotelina 1 no solo genera expresión de eNOS e iNOS, sino que también favorece Migración de Monocitos que igualmente expresan iNOS y Hemoxigenasa - 1 favoreciendo la Producción de Monóxido de Carbono y mayor Vasodilatación. En condiciones normales, el eritrocito con un tamaño de 7-8 µm atraviesa capilares pulmonares de pequeño diámetro (8-15 µm), y está en contacto estrecho con la membrana alvéolo capilar; allí las células eritroides circulan "en fila", cargando sus moléculas de hemoglobina con oxígeno. pequeños cambios en las resistencias pre y/o post capilares generan modificaciones pasivas en el diámetro del capilar alveolar; el incremento del diámetro capilar por sobreproducción de moléculas dilatadoras (ON) conlleva a un aumento en el flujo sanguíneo a nivel local; de esta manera se altera la mecánica de oxigenación y se produce hipoxemia como resultado del "desajuste" en la ventilación/Perfusión (exceso de perfusión para una determinada ventilación),la alteración en la difusión/perfusión, debido a un incremento en las distancias de difusión del O2 hacia los eritrocitos por los capilares dilatados y verdaderos "shunts", anatómicos intrapulmonares (Figura 1) (18).
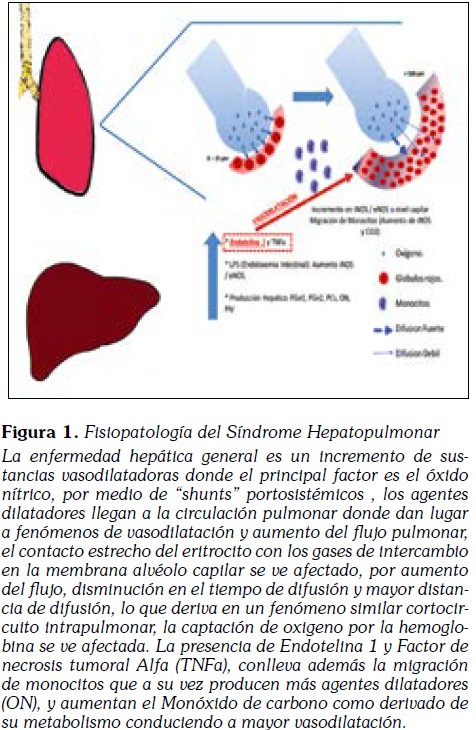
Por otro lado, el aumento de la frecuencia cardiaca presente en los pacientes con enfermedad hepática, genera menor tiempo de difusión a nivel de la membrana alveolo capilar, conduciendo a menor captura de oxigeno por parte del eritrocito circulante (19).
Manifestaciones Clínicas
La disnea con el ejercicio es la manifestación clínica más común, usualmente aparece después de trascurridos varios años de enfermedad hepática; el trastorno se acompaña además de platipnea y ortodisoxia, la cual hace referencia a un descenso del 5% o > de 4 mmHg en las PaO2 con cambios de posición supina a bipedestación (20).
La disnea también puede ser el resultado de la compresión del parénquima pulmonar por ascitis o efusión pleural, y/o otras enfermedades concomitantes que afecten el pulmón y el hígado (21). Se debe aclarar que a pesar de que la disnea es un síntoma usual en la cirrosis, la presencia de hipoxemia severa (PaO2 < 60 mmHg) no lo es y su hallazgo en ausencia de enfermedad cardiopulmonar previa es indicativo de la presencia de SHP (22).
Los signos clínicos presentes en el SHP no son específicos, incluyen teleangiectasias, acropaquia y Cianosis. Algunos autores resaltan que las teleangiectasias son las manifestaciones dérmicas del SHP, esto justificado en el hallazgo de mayor hipoxemia en pacientes cirróticos con esta manifestación comparados con los que no la tienen, así como en la presencia de patrones fisiopatológicos similares, basados en el desequilibrio entre agentes vasoconstrictores y vasodilatadores (23).
Diagnóstico
El diagnostico parte de la evidencia de alteración en la oxigenación en el contexto de dilatación vascular pulmonar y enfermedad hepática o hipertensión portal. En estadios tempranos no es necesario documentar hipoxemia, la altercación inicial es un descenso de la PaCO2 a causa de hiperventilación. La forma apropiada de evaluar las alteraciones a nivel de oxigenación es mediante el cálculo de la diferencia Alveolo - Arterial de Oxigeno (A-aPO2), cuyo punto de corte es de 15 mmHg para menores de 65 años y 20 mmHg para mayores de 65 años (24).
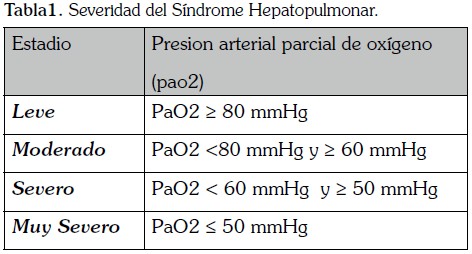
La evaluación en conjunto de las PaO2 y la A-aPO2, clasifica el síndrome en cuatro estadios de severidad (25) (Tabla 1)
En el abordaje diagnóstico, la evaluación de la oximetría tiene especial interés, según estudio realizado por Arguedas M, et al, (26) un valor de oximetría menor de 94%, se relaciona de manera confiable con una PaO2 < 60 mmHg (sensibilidad: 100%, Especificidad: 93%, CI (95): 0.97 - 1.0). La utilidad del hallazgo radica en el impacto de los valores bajos de PaO2 en la mortalidad, dado que todos los pacientes con PaO2 < 60 mmHg, tienen indicación de angiografía pulmonar, para evaluar malformaciones arteriovenosas susceptibles de embolización y/o priorización para trasplante hepático (27).
Un estudio reciente, realizado por Gupta, et al, ha cuestionado la utilidad de una medición aislada de PaO2, ya que una segunda evaluación de la PaO2 puede cambiar la gravedad del SHP o incluso descartarlo. En este trabajo la medición de PaO2 en dos periodos diferentes permite una mejor aproximación al diagnóstico de SHP. Durante el estudio entre el 8.6% y el 15% de los pacientes que fueron diagnosticados con SHP en una primera medición de PaO2, no tenían la enfermedad tras una segunda medición; sin embargo, los pacientes que tienen un valor inicial de PaO2 < 60 mmHg, no requieren de una segunda medición (28).
La evaluación de la dilatación de los capilares pulmonares tiene diversas opciones, los hallazgos de la radiografía simple son muy poco específicos, en el mejor de los casos se puede llegar a apreciar cambios intersticiales secundarios a la dilatación de los pequeños vasos pulmonares (16).
El ecocardiograma con solución salina agitada es un método ampliamente utilizado, muy sensible y poco específico, es positivo en 34% al 47% de los pacientes con Cirrosis (29). El método se basa en la presencia de microburbujas en la aurícula derecha tras la administración endovenosa periférica de solución salina agitada, las cuales se aprecian 3 a 6 ciclos cardiacos después en la aurícula izquierda indicando paso de la mismas por vasculatura anormalmente dilatada (22).
El tiempo de paso de las microburbujas del corazón derecho al izquierdo permite diferenciar la dilatación vascular intrapulmonar del SHP de las comunicaciones interauriculares e interventriculares, ya que en el shunt intracardiaco las burbujas aparecen en cavidades izquierdas en 1 a 2 ciclos. El uso de ecocardiograma trasesofágico (ECO TE), es mucho más sensible y específico ya que mediante sus proyecciones es posible ver las microburbujas en las venas pulmonares (30).
La gammagrafía de perfusión pulmonar con partículas de albumina marcada, es otro de los métodos empleados para demostrar la presencia de vasodilatación pulmonar, el estudio se basa en la administración de partículas de albumina macroagregadas marcadas con Tc99 (diámetro de 10 - 90 µm) las cuales, por su tamaño, en condiciones normales son atrapadas en más de un 90% en el pulmón. En pacientes con SHP, la dilatación capilar pulmonar permite el paso de estas partículas hacia la circulación sistémica depositándose en cerebro, riñón y tiroides. El porcentaje de captación en tejido cerebral es medido y con base en el mismo, se aplica una fórmula matemática para medir el porcentaje del "shunt" (31) . La gasimetría también es de utilidad para la evaluación del "Shunt", las mediciones se realizan inicialmente con fracción inspirada de 21% y posteriormente con fracción inspirada de Oxígeno de 100%, si la PaO2 es menor de 200 mmHg, se debe sospechar la presencia de dilataciones vasculares intrapulmonares Severas, comunicación AV discreta o "Shunt" Intracardiaco (16,20).
La evaluación de la disfunción hepática o la hipertensión portal, se hace mediante estudios séricos de marcadores de daño hepático, imagenología, diagnostico por histopatología y medición de presiones portales (28).
Recientemente se ha estudiado el valor del antígeno del Factor de Von Willebrand (vWB Ag) en la detección de síndrome Hepatopulmonar. Un estudio realizado por Horvatitis, et al, evaluó la presencia de estos antígenos en pacientes con daño vascular, demostrando este como un factor independiente para el desarrollo de SHP. Para un punto de corte de 328%, con sensibilidad del 100% y especificidad de 53.5%, con VPP de 36.9% y VPN: 100% (32).
Tratamiento
El SHP, es una indicación de trasplante hepático ortotópico independientemente de la severidad de la hipoxemia, éste es de carácter curativo; según las series de Mayo Clinic, la sobrevida de los pacientes trasplantados a 5 años es del 76% (16).
La presencia de una PaO2 < 50 mmHg se asocia con altas tasas de mortalidad por lo tanto los pacientes con PaO2 < 60 mmHg deben ser trasplantados lo más pronto posible (33, 34).
Basado en los hallazgos fisiopatológicos del SHP, se han estudiado múltiples opciones terapéuticas (Tabla 2), todas ellas con plausibilidad biológica, pero con resultados desalentadores, lo que hace que el trasplante ortotópico, se siga manteniendo como el tratamiento Ideal (35).
Hipertensión Portopulmonar (HPP)
Inicialmente descrita en 1951 por Mantz y Craige (36), es una indicación de trasplante ortotópico de hígado independiente de la severidad de la disfunción hepática. Esta condición se clasifica dentro de la causa de Hipertensión Pulmonar del grupo I según OMS (37) con una prevalencia del 4.5% al 10% (38).
La HPP se define como el aumento de la presión arterial pulmonar media, asociada a un aumento de la resistencia vascular pulmonar e hipertensión portal en un paciente con o sin enfermedad Hepática avanzada. Se estima que hasta un 10% de los casos puede presentarse a causa de Hipertensión portal sin alteración hepática estructural (especialmente para casos de infección por Schistosoma mansoni) (39).
El principal Factor de riesgo para el desarrollo de HPP es la presencia de hepatitis autoinmune (4.02, 95% CI 1.14-14.23, P <0.031) seguido de género femenino (OR 2.90, 95% IC 1.20-7.01, P < 0.018), por el contrario, la Infección por virus de hepatitis C confiere un bajo riesgo (OR: 0.24, 95% CI 0.09-0.65, P <0.005) (40).
La sobrevida de los pacientes con Hipertensión Porto-pulomonar es cercana a los 15 meses, estos pacientes tienen además 3 veces más riesgo de morir respecto a otros pacientes con causas diferentes de Hipertensión Arterial Pulmonar (41).
Fisiopatología
La HPP se manifiesta con un engrosamiento de la capa media del capilar pulmonar, acompañado de estrechez y obliteración de la luz vascular por fibrosis de la íntima, proceso que se ha denominado remodelación (42). Histopatológicamente, hay un predominio de proliferación de células endoteliales, desarrollo de lesiones plexiformes, necrosis fibrinoide y extensión de capa muscular en arteriolas pulmonares (que en condición normal no tiene capa muscular) (43).
El mecanismo molecular del HPP no está completamente definido, se cree que se origina de un estado hiperdinámico secundario a la enfermedad hepática, que produce aumento del flujo a nivel pulmonar, generando fuerzas de fricción capilar con respuesta de vasoconstricción, aumento las resistencias vasculares y remodelación vascular progresiva (44).
Esta condición se caracteriza por un desequilibrio, entre sustancias vasoactivas, existe un Incremento de la Endotelina 1, producida por las células endoteliales y que en presencia de "shunts" porto sistémicos en pacientes con hipertensión portal se puede fugar hacia la Circulación Pulmonar, generando procesos de remodelación Vascular (45).
El ON, por su parte tiene una producción sinusoidal disminuida e insuficiente para corregir los aumentos de presión en el sistema venoso portal; esto además tiene un efecto directo en las plaquetas promoviendo su agregación y formación de trombos (42).
Manifestaciones Clínicas
Este trastorno es inicialmente asintomático, el síntoma más común es la disnea con el ejercicio, con menor frecuencia los pacientes pueden experimentar fatiga, palpitaciones, sincope o dolor torácico (46).
Los hallazgos del examen físico son dependientes de la gravedad y pueden incluir reforzamiento del segundo ruido, soplos sistólicos sugestivos de insuficiencia tricuspidea, edema de miembros inferiores, ingurgitación yugular o ascitis, estos últimos que pueden ser el resultado de cirrosis descompensada o falla cardiaca derecha (47).
Diagnóstico
La evaluación de la Hipertensión Arterial Pulmonar (HTP), puede realizarse mediante ecocardiografía transtorácica toda vez que exista gradiente transvalvular tricuspideo. Este método diagnostico cuenta con alta sensibilidad (97%) y moderada especificidad (77%) pudiendo sobrestimar los valores. Su mayor utilidad radica en la posibilidad de excluir el diagnostico con presiones medias de la arteria pulmonar calculada en menos de 15 mmHg (48).
El cateterismo derecho es el "Gold Standard" para el diagnóstico, mediante el mismo es posible medir directamente las presiones medias de la arteria pulmonar, presión de cuña pulmonar y resistencia arterial pulmonar. El estudio se indica en pacientes con presiones sistólicas de la arteria pulmonar mayores o iguales a 50 mmHg medidas por ecocardiografía (16,49).
El Diagnóstico definitivo se establece con un valor de PmAP ≥ de 25 mmHg en reposo Y/0 ≥ 30 mmHg con el ejercicio, presión de Cuña < de 15 mmHg y resistencia Vascular pulmonar (RVP) mayor de 240 dinas/s/Cm (50). Con base en la medición de la PmAP la hipertensión portopulmonar se clasifica en tres grados (Tabla 3).
Otros hallazgos sugestivos para el diagnóstico, son los signos de hipertensión pulmonar precapilar en radiografía de tórax y los signos indirectos de sobrecarga ventricular derecha en electrocardiograma (Bloqueo de rama derecha, desviación derecha del eje, e inversión de Onda T en derivadas anteriores).
Tratamiento
La evidencia de tratamiento farmacológico de la Hipertensión Portopulmonar es limitada, su clasificación dentro de los cuadros de Hipertensión Pulmonar del grupo I, permite el uso de medicamentos vasodilatadores: Prostanoides, Inhibidores de la Endotelina 1, e inhibidores de la Fosfodiesterasa.
La decisión de tratamiento y el tipo de tratamiento se fundamenta en la severidad del cuadro; en procesos leve (PsAP < 35 mmHg), el tratamiento no es obligatorio, dado que algunos estudios han demostrado que el resultado al trasplante hepático, no difiere respecto a aquellos paciente sin Hipertensión Portopulnar (39).
El uso de vasodilatadores para control de PmAp ha demostrado se mejor respecto a la comparación con grupos de pacientes sin tratamiento, estudios realizados al respecto, han demostrado una sobrevida a 5 años de 45% en aquellos que emplearon un fármaco especifico, incrementable hasta 76% para quienes además del fármaco fueron llevados a trasplante Hepático (51).
Dentro de los agentes farmacológicos, los análogos prostanoides han mostrado un mejor, rendimiento, así se observó en un estudio realizado con Epoprostenol endovenoso donde se demostraron cambios agudos de disminución en la resistencia vascular pulmonar (-34% ± 18%), presión media de la arteria pulmonar (-16% ± 10%), y aumento en el gasto cardiaco (+21% ±18%) (P <0.05), así como un beneficio sostenido a largo plazo (52). Su efectividad esta contrarrestada por la presencia de efectos adversos y pobre tolerabilidad (dolor articular, diarrea, eritema, estados de alto gasto cariaco, infección trombosis).
Otros grupos farmacológicos incluyen, Inhibidores del fosfodiesterasa dentro de ellos el Sildenafil, el cual ha demostrado inhibir el crecimiento del musculo liso vascular pulmonar, disminuyendo la RVP de 398 a 179 dinas.sec.cm5 y la PmAP de 56 a 28- 31 mmHg, con mejoría en caminata de 6 minutos, tras cuatro semanas de tratamiento (53). Su uso debe ser cauteloso dado que causa vasodilatación esplácnica con subsecuente Incremento en la presión Portal (54).
Los Inhibidores de la Endotelina 1, Bosentan y Ambrisentan bloquean la respuesta de vasoconstricción inducida por la Endotelina y conllevan a una mejoría en caminata de 6 minutos, Aumento de Índice Cardiaco, reducción de la Resistencia Vascular pulmonar y mejoría sintomática (54).
Estudios realizados por Hoeper et al, evaluaron la supervivencia a 1, 3 y 5 años de pacientes tratados con Bosentan versus análogo prostanoide (iloprost), encontrando que esta fue de 94%, 89%, 89% VS 72%, 62%, 64% respectivamente, documentaron además un disminución neta de la RVP de 345±361 dynes.sec.cm5 (55).
El trasplante Hepático, ha mostrado tener utilidad, en pacientes con PmAp ≤ 35 mmHg y resistencias vasculares Pulmonares menores de 240 dinas*s*Cm5, la mortalidad en estos casos es de 0% . Valores superiores a estos se asocian a un incremento en la mortalidad, la cual es de 50% para pacientes con PmAP > 35 / <45 mmHg y de 100% para aquellos con PmAP ≥ 45 mmHg (56).
Lo anterior sugiere que hay un beneficio en el tratamiento pre trasplante de los pacientes con Hipertensión Portopulmonar moderada a severa. Este beneficio fue observado por Ashfat el at, quien encontró una sobrevida de 90.9%, 80.8% y 67.3%, a 1, 2 y 5 años respectivamente, en pacientes con diagnóstico de Hipertensión Portopulmonar, con PmAP > 35 mmHg y que fueron pre tratados con Epoprostenol hasta alcanzar valores inferiores a ésta cifra (57).
El pronóstico de la hipertensión portopulmonar sin tratamiento es malo, la sobrevida a un año es de 85%, con los tres años de 38% a 68%, y a cinco años de solo 40% (58).
Hidrotórax Hepático (HH)
Se define como derrame pleural usualmente mayor a 500 cc, de predominio en hemitorax derecho, no asociado a condición cardiaca, pulmonar o pleural primaria, en un paciente con cirrosis de cualquier causa; su prevalencia es del 5% al 12% (59).
Las descripciones iniciales y parte del sustento fisiopatológico se acercan al año de 1955 donde se establece por primera vez la presencia de una fenestración diafragmática en un paciente con fibrosis y derrame pleural (60).
Fisiopatología
Se han propuesto múltiples mecanismos fisiopatológicos, la hipoalbuminemia, el aumento de flujos y presiones en el conducto torácico y la vena ácigos son algunas de las alteraciones presentes en los pacientes con cirrosis que pueden estar en relación con el desarrollo de Hidrotórax Hepático (61).
Estudios posteriores demostraron la presencia de alteraciones diafragmáticas que se asocian a paso directo del líquido ascítico a la cavidad pleural. La infusión de aire intraperitoneal, en pacientes con esta condición permite detectar el paso del mismo a la cavidad torácica en imágenes de Rayos X tomadas 48 Horas después (62). Esta teoría explica el porqué del predomino derecho si se tenía en cuenta que por el desarrollo embriológico el hemidiafragma derecho es más propenso a tener defectos (59).
En los pacientes cirróticos el aumento en la presión Intrabdominal por valsalva, ascitis, distensión, genera la formación de pequeñas ampollas del peritoneo al protruir sobre los defectos diafragmáticos estos se rompen hacia la cavidad pleural dado lugar al desarrollo del derrame pleural cuando la capacidad de absorción de la pleura es sobrepasada. La presión intratoracica negativa generada durante la inspiración favorece el ascenso del líquido (63).
Manifestaciones Clínicas
Los pacientes inicialmente se presentan con dificultad respiratoria en el ejercicio, en la medida que el fluido se va acumulando se puede presentar dolor pleurítico, ortopnea, tos e incluso progresión a falla ventilatoria (64).
La ascitis es detectable hasta en un 80% de los Casos de Hidrotórax Hepático, sin embargo, su ausencia no excluye el diagnóstico (65).
En pacientes que desarrollan peritonitis bacteriana espontánea, el líquido pleural puede tornarse turbio y de características Infecciosas (tabla 4). Sin embargo, el empiema bacteriano espontaneo puede aparecer en ausencia de peritonitis bacteriana espontánea (61).
Diagnóstico
La historia médica de cirrosis y el hallazgo radiográfico de derrame pleural constituyen el primer paso en el diagnóstico; la ubicación del derrame pleural, tiene la siguiente distribución: 85% derecho, 13% izquierdo, 2%bilateral (66). El hallazgo requiere de toracentesis para excluir otras causas; el estudio del líquido pleural, revela un líquido exudativo, cuyas características se resaltan en la tabla 4.
Solo en el 18% de los pacientes cirróticos con derrame pleural, éste es explicado por una causa distinta al HH (59). La identificación de comunicación entre las cavidades de espacio pleural, es una herramienta útil para el diagnóstico, esto se realiza mediante la inyección intraperitoneal de aire o agentes de contraste (Albumina marcada con Tc99 o Coloides de Sulfuro marcados con Tc99). Este estudio tiene una sensibilidad del 71% y especificidad del 100% (61).
Tratamiento
El uso de dieta baja en sal y con restricción hídrica constituye la base del tratamiento, sin embargo, estas medidas no suelen ser suficientes, otras opciones incluyen toracentesis evacuatoria, pleurodesis, y Consideración de Shunt Porto-sistémico.
La toracentesis evacuatoria es una opción en casos sintomáticos, el volumen extraído por procedimiento se sugiere que no sea mayor a 2 litros, considerado el bajo volumen, administración de albumina post procedimiento no es necesario (67).
La inserción de tubos para drenaje no es recomendada en estos pacientes, aun cuando se identifique pleuritis bacteriana espontanea; solo en casos en los que halla presencia de empiema se indicara el uso de tubo de toracostomía para drenaje. Los drenajes prolongados dan cabida a la perdida de electrolitos y proteínas, con desarrollo de enfermedad renal y alteraciones inmunológicas como secuelas (68).
La pleurodesis química, es poco efectiva, dado a la dificultad para el sellado químico de las pleuras por la rápida acumulación de líquido en el espacio pleural (69). Adicionalmente, el procedimiento tiene complicaciones significativas que incluyen lesión renal aguda, neumonía, neumotórax y encefalopatía con una mortalidad reportada del 27% a los 30 días (70).
Se estima que cerca de un 20% de los casos son refractarios al tratamiento (71), en estas situaciones la cirugía asistida por videotoracoscopia para cierre de defectos diafragmáticos con o sin pleurodesis y la realización de shunt postosistémico son las terapias de elección. El primero de ellos aun con falta de datos que apoyen su uso y pequeños grupos de casos que indican una tasa de éxito del 63% al 85% (72).
El shunt portosistémicos es una medida efectiva, su fundamento se basa en la disminución de la presión portal, y la filtración hacia el espacio peritoneal, descendiendo hasta niveles que pueden ser fácilmente drenados por el sistema linfático. De esta forma a diferencia de los diuréticos y la paracentesis, este procedimiento contrarresta la causa que conlleva a la ascitis (73). La evidencia indica que este procedimiento es efectivo con tasa de respuesta completa de 65%, y sobrevida a un año entre 48% y 64% (74).
Conclusión
La cirrosis es una enfermedad que se relaciona con la disfunción de múltiples órganos, su comportamiento conduce a un rápido deterioro con pobres tasa de sobrevida.
El compromiso pulmonar en la cirrosis, es una condición frecuente, con tres presentaciones que incluso pudiesen sobreponerse, convirtiéndose en un factor independiente de mortalidad, e indicación de trasplante, su diagnóstico depende inicialmente de la aceptación de la existencia de estas condiciones y su búsqueda activa en los pacientes con enfermedad hepática. Las opciones de tratamiento son diversas y algunas de ellas mejoran considerablemente la calidad y tasa de sobrevida.
Conflicto de intereses
Los autores declaran no tener de manera directa o indirecta ningún tipo de conflicto de intereses financieros, académicos o laborales que puedan poner en peligro la validez de este estudio.Referencias
1. Moller, S., et al., Pathophysiological aspects of pulmonary complications of cirrhosis. Scand J Gastroenterol, 2007. 42(4): p. 419-27. [ Links ]
2. Swanson, K.L., R.H. Wiesner, and M.J. Krowka, Natural history of hepatopulmonary syndrome: Impact of liver transplantation. Hepatology, 2005. 41(5): p. 1122-9. [ Links ]
3. D'Amico, G., G. Garcia-Tsao, and L. Pagliaro, Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol, 2006. 44(1): p. 217-31. [ Links ]
4. Rehm, J., A.V. Samokhvalov, and K.D. Shield, Global burden of alcoholic liver diseases. J Hepatol, 2013. 59(1): p. 160-8. [ Links ]
5. Scaglione, S., et al., The Epidemiology of Cirrhosis in the United States: A Population-based Study. J Clin Gastro enterol, 2014. [ Links ]
6. Sureka, B., et al., Thoracic perspective revisited in chronic liver disease. Gastroenterol Rep (Oxf), 2015. [ Links ]
7. Montoya, A., et al., Caracterización epidemiológica de pacientes con cirrosis en una consulta de gastroenterología en Pereira, Colombia, 2009-2012. Rev Méd Risaralda, 2014. 20(2): p. 86-94. [ Links ]
8. Machicao, V.I., M. Balakrishnan, and M.B. Fallon, Pulmonary complications in chronic liver disease. Hepatology, 2014. 59(4): p. 1627-37. [ Links ]
9. King, P.D., R. Rumbaut, and C. Sanchez, Pulmonary manifestations of chronic liver disease. Dig Dis, 1996. 14(2): p. 73-82. [ Links ]
10. Sen, D.S. and D.G.J. Alexander, Pulmonary complications in liver disease. British Journal of Hospital Medicine, 2009. 70(12): p. 685-691. [ Links ]
11. Kennedy, T.C. and R.J. Knudson, Exercise-aggravated hypoxemia and orthodeoxia in cirrhosis. Chest, 1977. 72(3): p. 305-9. [ Links ]
12. Raevens, S., et al., Hepatopulmonary syndrome and portopulmonary hypertension: recent knowledge in pathogenesis and overview of clinical assessment. Liver International, 2015. 35(6): p. 1646-1660. [ Links ]
13. Grace, J.A. and P.W. Angus, Hepatopulmonary syndrome: update on recent advances in pathophysiology, investigation, and treatment. J Gastroenterol Hepatol, 2013. 28(2): p. 213-9. [ Links ]
14. Rodríguez-Roisin, R. and M.J. Krowka, Hepatopulmonary Syndrome - A Liver-Induced Lung Vascular Disorder. New England Journal of Medicine, 2008. 358(22): p. 2378-2387. [ Links ]
15. Schenk, P., et al., Prognostic significance of the hepatopulmonary syndrome in patients with cirrhosis. Gastroenterology, 2003. 125(4): p. 1042-52. [ Links ]
16. Singh, C. and J.S. Sager, Pulmonary complications of cirrhosis. Med Clin North Am, 2009. 93(4): p. 871-83, viii. [ Links ]
17. Carter, E.P., et al., Regulation of heme oxygenase-1 by nitric oxide during hepatopulmonary syndrome. Am J Physiol Lung Cell Mol Physiol, 2002. 283(2): p. L346-53. [ Links ]
18. Zhang, H.-Y., et al., Intestinal endotoxemia plays a central role in development of hepatopulmonary syndrome in a cirrhotic rat model induced by multiple pathogenic factors. World Journal of Gastroenterology : WJG, 2007. 13(47): p. 6385-6395. [ Links ]
19. Thorens, J.B. and A.F. Junod, Hypoxaemia and liver cirrhosis: a new argument in favour of a "diffusion-perfusion defect". Eur Respir J, 1992. 5(6): p. 754-6. [ Links ]
20. Rodriguez-Roisin, R., et al., Pulmonary-Hepatic vascular Disorders (PHD). Eur Respir J, 2004. 24(5): p. 861-80. [ Links ]
21. Mohammad Alizadeh, A.H., et al., Clinical features of hepatopulmonary syndrome in cirrhotic patients. World J Gastroenterol, 2006. 12(12): p. 1954-6. [ Links ]
22. Lv, Y. and D. Fan, Hepatopulmonary Syndrome. Digestive Diseases and Sciences, 2015. 60(7): p. 1914-1923. [ Links ]
23. Silverio Ade, O., et al., Are the spider angiomas skin markers of hepatopulmonary syndrome? Arq Gastroenterol, 2013. 50(3): p. 175-9. [ Links ]
24. Koch, D.G. and M.B. Fallon, Hepatopulmonary syndrome. Clin Liver Dis, 2014. 18(2): p. 407-20. [ Links ]
25. Pascasio, J.M., et al., Prevalence and Severity of Hepatopulmonary Syndrome and Its Influence on Survival in Cirrhotic Patients Evaluated for Liver Transplantation. American Journal of Transplantation, 2014. 14(6): p. 1391-1399. [ Links ]
26. Arguedas, M.R., et al., Utility of pulse oximetry screening for hepatopulmonary syndrome. Clin Gastroenterol Hepatol, 2007. 5(6): p. 749-54. [ Links ]
27. Poterucha, J.J., et al., Failure of hepatopulmonary syndrome to resolve after liver transplantation and successful treatment with embolotherapy. Hepatology, 1995. 21(1): p. 96-100. 28. [ Links ]
28. Gupta, S., D. Nayyar, and G. Pomier-Layrargues, Variability of oxygenation in possible hepatopulmonary syndrome: effects of requiring two abnormal arterial blood gas results for diagnosis. Dig Dis Sci, 2015. 60(6): p. 1848-55. [ Links ]
29. Hopkins, W.E., A.D. Waggoner, and B. Barzilai, Frequency and significance of intrapulmonary right-to-left shunting in end-stage hepatic disease. Am J Cardiol, 1992. 70(4): p. 516-9. [ Links ]
30. Rollan, M.J., et al., Value of contrast echocardiography for the diagnosis of hepatopulmonary syndrome. Eur J Echocardiogr, 2007. 8(5): p. 408-10. [ Links ]
31. Surasi, D., P. Manapragada, and P. Bhambhvani, Lung perfusion imaging in hepatopulmonary syndrome using 99mTc macroaggregated albumin. Journal of Nuclear Cardiology, 2015. 22(3): p. 586-588. [ Links ]
32. Horvatits, T., et al., Von Willebrand factor antigen for detection of hepatopulmonary syndrome in patients with cirrhosis. J Hepatol, 2014. 61(3): p. 544-9. [ Links ]
33. Fallon, M.B., et al., Model for end-stage liver disease (MELD) exception for hepatopulmonary syndrome. Liver Transpl, 2006. 12(12 Suppl 3): p. S105-7. [ Links ]
34. Iyer, V.N., et al., Hepatopulmonary syndrome: favorable outcomes in the MELD exception era. Hepatology, 2013. 57(6): p. 2427-35. [ Links ]
35. Eshraghian, A., A.A.l. Kamyab, and S.K. Yoon, Pharmacological Treatment for Hepatopulmonary Syndrome. BioMed Research International, 2013. 2013: p. 670139. [ Links ]
36. Mantz, F.A., Jr. and E. Craige, Portal axis thrombosis with spontaneous portacaval shunt and resultant cor pulmonale. AMA Arch Pathol, 1951. 52(1): p. 91-7. [ Links ]
37. Simonneau, G., et al., Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol, 2013. 62(25 Suppl): p. D34-41. [ Links ]
38. Cartin-Ceba, R. and M.J. Krowka, Portopulmonary hypertension. Clin Liver Dis, 2014. 18(2): p. 421-38. [ Links ]
39. Aldenkortt, F., et al., Portopulmonary hypertension and hepatopulmonary syndrome. World Journal of Gastroenterology : WJG, 2014. 20(25): p. 8072-8081. [ Links ]
40. Kawut, S.M., et al., Clinical risk factors for portopulmonary hypertension. Hepatology, 2008. 48(1): p. 196-203. [ Links ]
41. Krowka, M.J., Portopulmonary hypertension and the issue of survival. Liver Transpl, 2005. 11(9): p. 1026-7. [ Links ]
42. Nayak, R., D. Li, and G. Matuschak, Portopulmonary hypertension. Current Gastroenterology Reports, 2009. 11(1): p. 56-63. [ Links ]
43. Dickinson, M.G., et al., The role of disturbed blood flow in the development of pulmonary arterial hypertension: lessons from preclinical animal models. Am J Physiol Lung Cell Mol Physiol, 2013. 305(1): p. L1-14. [ Links ]
44. Bosch, J., Vascular deterioration in cirrhosis: the big picture. J Clin Gastroenterol, 2007. 41 Suppl 3: p. S247-53. [ Links ]
45. Golbin, J.M. and M.J. Krowka, Portopulmonary hypertension. Clin Chest Med, 2007. 28(1): p. 203-18, ix. [ Links ]
46. Hoeper, M.M., M.J. Krowka, and C.P. Strassburg, Portopulmonary hypertension and hepatopulmonary syndrome. The Lancet, 2004. 363(9419): p. 1461-1468. [ Links ]
47. Ratti, L. and M. Pozzi, The pulmonary involvement in portal hypertension: portopulmonary hypertension and hepatopulmonary syndrome. Gastroenterol Hepatol, 2006. 29(1): p. 40-50. [ Links ]
48. Torregrosa, M., et al., Role of Doppler echocardiography in the assessment of portopulmonary hypertension in liver transplantation candidates. Transplantation, 2001. 71(4): p. 572-4. [ Links ]
49. Galiè, N., et al., Guidelines for the diagnosis and treatment of pulmonary hypertension. The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Vol. 30. 2009. 2493-2537. [ Links ]
50. Troy, P.J. and A.B. Waxman, Portopulmonary Hypertension: Challenges in Diagnosis and Management. Therapeutic Advances in Gastroenterology, 2009. 2(5): p. 281-286. [ Links ]
51. Swanson, K.L. and M.J. Krowka, Arterial oxygenation associated with portopulmonary hypertension. Chest, 2002. 121(6): p. 1869-75. [ Links ]
52. Krowka, M.J., et al., Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology, 1999. 30(3): p. 641-8. [ Links ]
53. Makisalo, H., et al., Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transpl, 2004. 10(7): p. 945-50. [ Links ]
54. Raevens, S., et al., Oral vasodilator therapy in patients with moderate to severe portopulmonary hypertension as a bridge to liver transplantation. Eur J Gastroenterol Hepatol, 2013. 25(4): p. 495-502. [ Links ]
55. Hoeper, M.M., et al., Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J, 2007. 30(6): p. 1096-102. [ Links ]
56. Krowka, M.J., et al., Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transpl, 2004. 10(2): p. 174-82. [ Links ]
57. Ashfaq, M., et al., The impact of treatment of portopulmonary hypertension on survival following liver transplantation. Am J Transplant, 2007. 7(5): p. 1258-64. [ Links ]
58. Krowka, M.J., et al., Portopulmonary hypertension: a report from the US-based REVEAL Registry. Chest, 2012. 141(4): p. 906-15. [ Links ]
59. Lazaridis, K.N., et al., Hepatic hydrothorax: pathogenesis, diagnosis, and management. Am J Med, 1999. 107(3): p. 262-7. [ Links ]
60. Emerson, P.A. and J.H. Davies, Hydrothorax complicating ascites. Lancet, 1955. 268(6862): p. 487-8. [ Links ]
61. Gur, C., Y. Ilan, and O. Shibolet, Hepatic hydrothorax-pathophysiology, diagnosis and treatment-review of the literature. Liver Int, 2004. 24(4): p. 281-4. [ Links ]
62. Lieberman, F.L., et al., Pathogenesis and treatment of hydrothorax complicating cirrhosis with ascites. Ann Intern Med, 1966. 64(2): p. 341-51. [ Links ]
63. Roussos, A., et al., Hepatic hydrothorax: Pathophysiology diagnosis and management. Journal of Gastroenterology and Hepatology, 2007. 22(9): p. 1388-1393. [ Links ]
64. Haas, A.R. and M.S. Machuzak, Hepatic hydrothorax: current approaches to diagnosis and therapy. Techniques in Gastrointestinal Endoscopy, 2009. 11(3): p. 114-117. [ Links ]
65. Benet, A., et al., Diagnosis of hepatic hydrothorax in the absence of ascites by intraperitoneal injection of 99m-Tc-Fluor colloid. Postgraduate Medical Journal, 1992. 68(796): p. 153-153. [ Links ]
66. Malagari, K., et al., Cirrhosis-related intrathoracic disease. Imaging features in 1038 patients. Hepatogastroenterology, 2005. 52(62): p. 558-62. [ Links ]
67. Krok, K.L., Hepatic hydrothorax: Current concepts. Clinical Liver Disease, 2014. 4(2): p. 35-37. [ Links ]
68. Borchardt, J., et al., Treating hepatic hydrothorax. BMJ : British Medical Journal, 2003. 326(7392): p. 751-752. [ Links ]
69. Rodriguez-Panadero, F. and V.B. Antony, Pleurodesis: state of the art. Eur Respir J, 1997. 10(7): p. 1648-54. [ Links ]
70. Lee, W.J., et al., Chemical pleurodesis for the management of refractory hepatic hydrothorax in patients with decompensated liver cirrhosis. Korean J Hepatol, 2011. 17(4): p. 292-8. [ Links ]
71. Siegerstetter, V., et al., Treatment of refractory hepatic hydrothorax with transjugular intrahepatic portosystemic shunt: long-term results in 40 patients. Eur J Gastroenterol Hepatol, 2001. 13(5): p. 529-34. [ Links ]
72. Cerfolio, R.J. and A.S. Bryant, Efficacy of video-assisted thoracoscopic surgery with talc pleurodesis for porous diaphragm syndrome in patients with refractory hepatic hydrothorax. Ann Thorac Surg, 2006. 82(2): p. 457-9. [ Links ]
73. Rossle, M. and A.L. Gerbes, TIPS for the treatment of refractory ascites, hepatorenal syndrome and hepatic hydrothorax: a critical update. Gut, 2010. 59(7): p. 988-1000. [ Links ]
74. Krishnamoorthy, T.L., M. Taneja, and P.E. Chang, Symptomatic hepatic hydrothorax successfully treated with transjugular intrahepatic portosystemic shunt (TIPS) -role of titration of portosystemic gradient reduction to avoid post TIPS encephalopathy. Clinical Case Reports, 2014. 2(3): p. 93-97. [ Links ]

