











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El síndrome de Leigh (SL), descrito por primera vez en 1951 por el neuropatólogo británico Denis Leigh, se presenta con una incidencia hasta de 1 en 40 000 recién nacidos vivos, sin predilección por género o raza y puede alcanzar una frecuencia hasta de 1:2000 RN en ciertas poblaciones como en Canadá 1,2. El SL es una enfermedad de origen mitocondrial de etiología genética, que ocasiona alteración en la producción energética. Clínicamente se describe un cuadro de retraso del neurodesarrollo o regresión de inicio agudo, con pérdida de habilidades adquiridas, que se acompaña de síntomas de tallo como estrabismo, nistagmo o trastorno de la deglución o síntomas piramidales, ataxia o falla ventilatoria. Las imágenes de resonancia magnética cerebral (RMC) tienen un papel importante en el diagnóstico de SL, donde se evidencian lesiones a nivel gangliobasal y tallo cerebral, y se encuentra elevación del pico de lactato en estudios de resonancia magnética con espectroscopia (RMS) 3,4.
A continuación, reportamos cinco casos con diagnóstico clínico y bioquímico del SL que ejemplifican la variabilidad clínica y gravedad encontrada en este grupo de pacientes, así como el abordaje diagnóstico ante la dificultad de acceso a pruebas moleculares confirmatorias.
Reporte de casos
Paciente 1
Adolescente femenina de 15 años producto del tercer embarazo a término, curso con alto riesgo por edad materna, padres no consanguíneos, con antecedentes de hermana fallecida a los 19 años con cuadro neurodegenerativo similar al cuadro clínico de la paciente, pero sin diagnóstico definitivo; no refiere antecedentes peri- o neonatales, al nacimiento normal, con adecuado peso y talla para la edad gestacional. Su neurodesarrollo fue normal hasta los 9 años; inicia con alteración en la marcha y debilidad en el hemicuerpo izquierdo, no hay otros antecedentes de importancia. Al momento de la valoración paciente sin dismorfias, sin compromiso de pares craneanos, hiperreflexia músculo-tendinosa generalizada, clonus bilateral y debilidad en hemicuerpo derecho, marcha hemiparética, movimientos coreicos en mano derecha, resto de examen neurológico normal. Se realiza RMC con lesión a nivel de núcleo caudado y lenticular bilateral simétrico por lo cual se amplían estudios con CPK, LDH y TSH normal, ceruloplasmina y cobre en orina normal, ácidos orgánicos en orina normales; pi-ruvato 0,175 (VR 0,030-0,100 mmol/l), lactato 0,659 mmol/l (VR [valor de referencia] 0,2-2,2 mmol/l) con una relación lactato/piruvato (L/P) 4.66 (VR- 0-25).
A los 11 años se realiza nueva RMC que evidencia compromiso del putamen bilateral a nivel mesencefálico y de los lóbulos frontales, aumento en la intensidad de la señal en T1 y T2 por lo que se realiza relación L/P en líquido cefalorraquídeo (LCR) en 34,22 (VR 0-25). Se confirma SL con espectroscopia por resonancia magnética computarizada (RMC) que evidencia elevación del ácido láctico (en el núcleo caudado y putamen). Se inicia manejo con carnitina, coenzima Q10, biotina, complejo B y zinc. La última valoración se realiza a los 17 años, no se encuentra deterioro clínico ni imagenológico.
Paciente 2
Lactante mayor masculino, de 15 meses de edad, producto del cuarto embarazo de padres jóvenes no consanguíneos, sin antecedentes familiares, período perinatal normal, nacimiento con adecuado peso para la edad gestacional, desarrollo psicomotor normal hasta los 10 meses cuando presenta pérdida de habilidades adquiridas del lenguaje, hipotonía, pérdida del sostén cefálico, tronco y presencia de distonías.
En la valoración inicial no se observan facies dismórficas, no establece contacto con el examinador, no fija y no sigue con la mirada, se encuentra hipertonía generalizada, posición de opistótonos e hiperreflexia músculo tendinosa. Se realizan estudios complementarios con amonio normal y ácido láctico levemente elevado (dato numérico no registrado en la historia clínica), la RMC a los 14 meses presenta hiperintensidad en T2 en núcleos lenticulares, tálamos y compromiso parcial de núcleo caudado bilateral, lesiones de aumento de la intensidad de la señal en T2 en el tallo cerebral potenciales evocados visuales y auditivos normales. Se realiza estudio de ácido láctico sanguíneo 5,36 mmol/l (VR 0,2-2), piruvato: 0,17 mmol/l (VR 0,03-0,12), relación L/P: 31 (VR: 0-25). Se inicia manejo con coenzima Q10, L-carnitina, y vitaminas: tiamina + riboflavina + vitamina B6 + fola-to+ vitamina B12 + biotina + ácido pantoténico.
A los 26 meses presenta convulsiones con adecuada respuesta a tratamiento con fenobarbital, continúa con posturas distónicas, sin mejoría en el desarrollo. El control de RMS demuestra compromiso de las regioneses gangliobasal, talámica y pontomesencefálica con disminución de N-acetil-aspartato y presencia de doble pico de lactato.
Paciente 3
Escolar femenina de 10 años; sin antecedentes perinatales ni familiares; padres no consanguíneos; producto del segundo embarazo de curso normal; a término; sin antecedentes perinatales de importancia; peso, talla y perímetro cefálico al nacimiento normales.
A los 2 años se evidencia aumento del tono muscular en forma progresiva y presencia de movimientos distónicos generalizados que fueron progresando hasta imposibilidad para la marcha, se acompaña de trastorno del lenguaje expresivo y discapacidad cognitiva leve. Al examen se encuentra con retardo pondoestatural, perímetro cefálico normal, palidez del disco óptico bilateral e hipertonía y distonía generalizadas. En los exámenes de laboratorio se registró elevación de lactato en plasma en valores entre 6 y 8 mmol/l (VR 0,2-2) y relación L/P: 33 (VR: 0-25). En la resonancia magnética cerebral (RMC) se evidencia lesiones hiperintensas en T2 en ganglios basales, putamen y núcleo caudado; en la espectroscopia por resonancia magnética (MRS) se observa elevación del pico de ácido láctico en la región ganglio basal bilateral. El perfil de carnitinas, cromatografía de ácidos orgánicos en orina, ceruloplasmina, cobre sérico, potenciales evocados auditivos y visuales fueron normales. Se realizó estudio molecular para déficit de pantotenato kinasa que fue negativo. Se inicia manejo con L-carnitina, vitamina B 12, biotina, tiamina y coenzima Q.
El cuadro clínico ha sido de progresión lenta, dependiente para la marcha y actividades de la vida cotidiana. Se encuentra en manejo interdisciplinario con terapias integrales.
Paciente 4
Paciente femenino de 4 años, sin antecedentes familiares o perinatales; producto del primer embarazo de padres consanguíneos de curso normal, parto a término normal; a partir de los 12 meses de vida inicia regresión en el desarrollo, con presencia de movimientos distónicos y coreicos asociados a pérdida de habilidades cognitivas y del lenguaje.
Se inician estudios con RMC con evidencia de lesiones hiperintensas en T2 en ganglios basales (núcleo caudado y putamen) y tallo cerebral; en líquido cefalorraquídeo (LCR) presenta elevación de ácido láctico: 6 mmol/L (VR 0,2-2), con relación L/P: 28 (VR 0-25). Los potenciales visuales y auditivos fueron normales, lo mismo que la cromatografía de aminoácidos en orina y plasma. En el momento paciente con microcefalia, retardo pondoestatural, trastorno de deglución, retraso severo en el desarrollo psicomotor, palidez del disco óptico bilateral, hiperreflexia músculo tendinosa generalizada, con cuadriparesia espástica con movimientos distónicos y coreoatetósicos en las extremidades. La MRS evidencia aumento del pico de ácido láctico en ganglios basales y tallo cerebral. Se inició manejo con L-carnitina, multivitaminas y coenzima Q. Por presencia de trastorno de la deglución y broncoaspiraciones a repetición, se realizó gastrostomía; ha recibido manejo interdisciplinario con terapias integrales. El cuadro clínico ha sido progresivo e involutivo y el paciente es dependiente en todas las actividades de la vida cotidiana.
Paciente 5
Prescolar masculino de 3 años, consulta por presencia de crisis convulsivas de 6 meses de evolución y regresión en el neurodesarrollo desde esa época, con pérdida del sostén cefálico y de tronco, posturas distónicas generalizadas y pérdida del lenguaje. Producto del primer embarazo de padres no consanguíneos, sin antecedentes familiares, con parto y período perinatal normal.
Durante el examen mostró pobre interacción con el examinador y pobre seguimiento visual y auditivo, se documenta hipertonía y distonía generalizadas, además mioclonías segmentarias con movimientos coreicos en las extremidades. Se realizan pruebas de ácido láctico en plasma: 4,316 mmol/l (VR 0,2-2), relación L/P en plasma 35 (VR: 0-25), ácido láctico en LCR 4,76 mmol/l (VR 0,2-2), gases arteriales: normales, cromatografía de aminoácidos en orina y plasma normal, cromatografía de ácidos orgánicos normal, neuroconducción y electro-miografía en las 4 extremidades normales; potenciales visuales con integridad de la vía retino cortical, auditivos: sugestivo de disfunción neurosensorial de la vía auditiva bilateral, video telemetría con presencia de brotes lentos irregulares sin actividad paroxística. La RMC muestra hiperintensidades en ganglios basales bilaterales con compromiso talámico parcial y extensión mesencefálica bilateral en la secuencia FLAIR y T2; en la MRS por resonancia magnética cerebral con presencia de aumento del pico del lactato en región ganglio-basal bilateral (núcleos putamen y caudado).
Muestra evolución desfavorable, requirió traqueotomía y gastrostomía, con movimientos distónicos generalizados y convulsiones de difícil control.
Discusión
Los casos presentados ilustran el amplio espectro clínico de la enfermedad con edades de presentación a partir de los 10 meses hasta los 15 años de vida. En la forma neonatal del SL, los síntomas son inespecíficos, suele presentarse con bajo peso para la edad gestacional, oligo-hidramnios, ventriculomegalia e incluso cardiomegalia; en ocasiones es posible encontrar en las neuroimágenes alteración en sustancia blanca 1. En la forma infantil, al igual que en la neonatal, los signos y síntomas iniciales son inespecíficos: episodios de vómito persistentes, hipotonía, falla del medro y retraso del neurodesarrollo, seguido de pérdida de las habilidades motoras y deterioro cognitivo de progresión variable; los cinco pacientes presentan deterioro cognitivo. Se pueden presentar otros síntomas neurológicos como espasticidad, distonía, convulsiones, ataxia, parálisis de pares craneanos, hipoacusia y disfagia 5,6. Teniendo en cuenta lo anterior, los cinco casos presentados corresponden a la forma infantil de la enfermedad y muestran el deterioro neurológico característico.
Se ha reportado hasta un 39 % de los pacientes con SL cursan con epilepsia, con presentación clínica y gravedad variables, secundaria a la heterogeneidad genética 1. Dos de los casos presentados presentaron epilepsia, uno de ellos con adecuada respuesta al tratamiento, mientras en el otro las crisis son de difícil control. En contraste, las distonías se presentaron como síntoma preponderante en este grupo de pacientes.
En el transcurso de la enfermedad es frecuente que se presenten exacerbaciones agudas o descompensaciones, las cuales están asociadas a momentos de estrés metabólico como infecciones o complicaciones respiratorias, con mayor pérdida de las habilidades adquiridas y empeoramiento de síntomas como ataxia, debilidad muscular generalizada o en músculos respiratorios 9; luego de estas descompensaciones es raro recuperar el desarrollo previamente alcanzado y cuando se logran periodos de estabilidad, estos tienen una duración variable 5,6. Dos de nuestros pacientes tienen una estabilidad clínica, otro ha progresado lentamente, otro presenta un cuadro desfavorable y otro ya se encuentra dependiente en todas sus actividades, lo que da un ejemplo de los estadios típicos de la enfermedad.
Aunque en los pacientes con SL (usualmente se ha descrito que cursan con compromiso multisistémico: síntomas cardiacos tales como cardiomiopatía hipertrófica, arritmias, alteraciones en la conducción; compromiso renal, hepatopatía, alteraciones hematológicas principalmente anemia y compromiso endocrinológico más frecuentemente diabetes y talla baja [5]), en los pacientes 3 y 4 se encontró retardo pondoestatural complicaciones descritas en la literatura. Los pacientes muestran hallazgos característicos en las neuroimágenes, lo que contribuye en gran medida al proceso diagnóstico 14,15 (tabla 1).
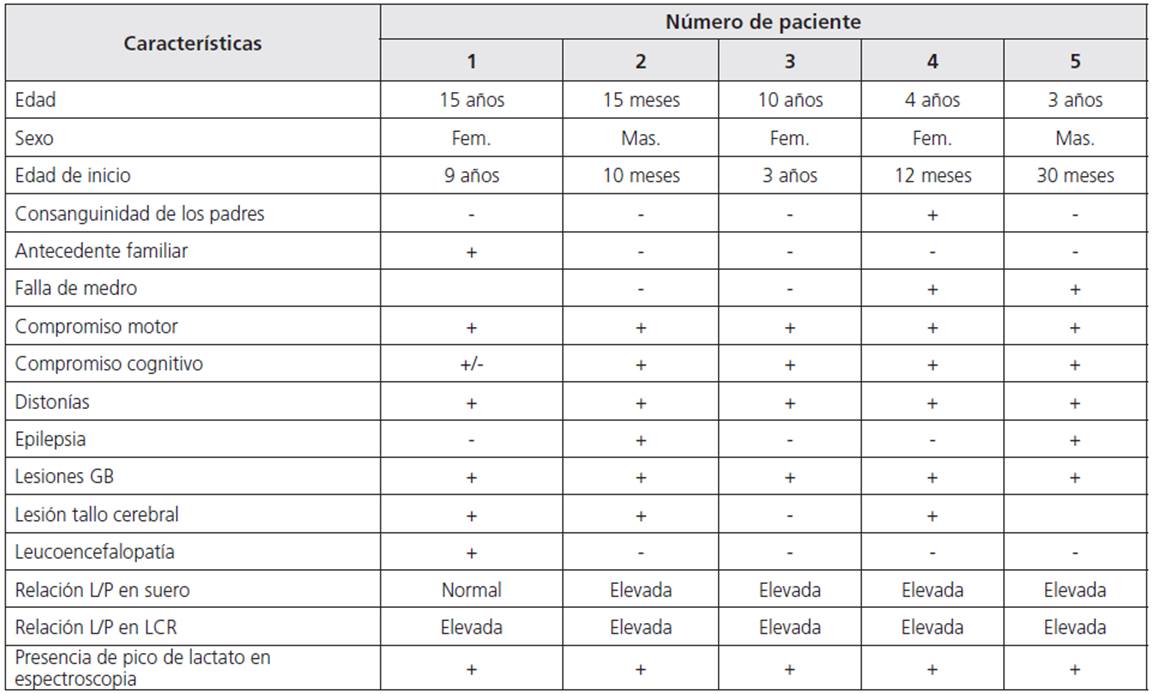
Bioquímicamente, la principal ayuda diagnóstica es la identificación de elevaciones del ácido láctico y su relación con piruvato en sangre y LCR. Estos metabolitos carecen de la sensibilidad y especificidad necesarias para considerarlos biomarcadores específicos de esta enfermedad, dado que hay varias causas fisiológicas y patológicas para su elevación, así como casos de pacientes en los cuales estos parámetros son normales 15-18. En los estudios realizados a los pacientes analizados, cuatro presentaron elevación de la relación L/P en plasma, con excepción del paciente 1, quien tuvo una relación normal en plasma y al realizarse el estudio en LCR presentó elevación significativa de ella, con lo que se pudo confirmar el diagnóstico inicial.
El diagnóstico confirmatorio de SL se realiza a través de la identificación de la mutación que lo causa. Hasta el momento se han descrito mutaciones en 60 genes, las cuales solo explican la mitad de los casos descritos. Estos genes son de origen tanto mitocondrial como nuclear y afectan principalmente el sistema de fosforilación oxidativa (OXPHOS), que lleva a cabo el proceso de producción energética a nivel mitocondrial 1,5,8,10 ,19) . En la tabla 2 se presentan las principales causas genéticas del SL y su frecuencia. En la actualidad, aunque las herramientas de diagnóstico molecular se pueden solicitar en Colombia, este examen no se procesa en el país, por lo que son de difícil accesibilidad, hecho que causa retrasos en el proceso diagnóstico. Adicionalmente, como toda herramienta diagnóstica, estas pruebas deben ser interpretadas en el contexto del paciente, los hallazgos de otros paraclínicos y los detalles técnicos de la realización de la prueba.
Tabla 2 Lesiones genéticas en la cadena respiratoria, asociadas con SL
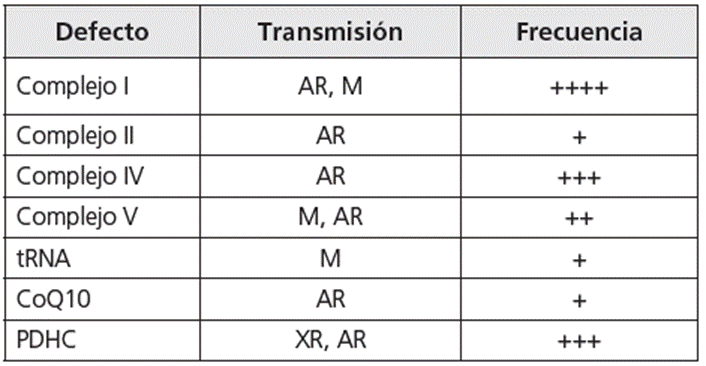
AR: autosómico recesivo; M: materno; XR: ligado a cromosoma X; PDHC: complejo piruvato deshidrogenasa; CoQ10: coenzima Q10.
Los casos presentados cumplen los criterios de diagnóstico definidos por Rahman en 1996; que establecen al SL como una enfermedad neurodegenerativa con compromiso motor y cognitivo, con signos o síntomas de compromiso en tallo cerebral o ganglios basales, asociado a niveles elevados de lactato en plasma o LCR (los valores pueden ser normales en el 25 % de los casos) y presencia de lesiones características en neuroimágenes por RMC o estudios anatomopatológicos 5,11,12. Los hallazgos en RMC se caracterizan por presencia de lesiones hi-perintensas en T2 simétricas, bilaterales principalmente en ganglios basales o tallo cerebral más específicamente en sustancia nigra, núcleo rojo y en médula o presencia de lesiones hipodensas en ganglios basales en la TAC 1,6,14. No obstante, las lesiones se pueden presentar en otras áreas típicas, como cordón espinal, tálamo y cerebelo 5,13. A nivel de RMC es posible identificar lesiones en sustancia blanca, que son confluentes, simétricas y bilaterales, y en ocasiones pueden ser confundidas con una leucodistrofia; estos pacientes pueden presentar lesiones tipo accidente cerebro vascular, atrofia cortical o cerebelosa y compromiso simétrico en núcleos olivares y dentado 14. Estudios complementarios con MRS cerebral, como los realizados en los pacientes presentados, permiten demostrar elevación de los picos de lactato tanto en tejido aparentemente normal como en parénquima afectado 4,15,16.
Todos los pacientes de este reporte presentaron compromiso imagenológico en ganglios basales y en uno de ellos se preservaron las estructuras en tallo cerebral (figura 1). El compromiso de sustancia blanca difusa de predominio frontal se presentó en uno de los pacientes en el que además se observó presencia del pico de lactato por espectroscopía en esta región y en los ganglios basales.
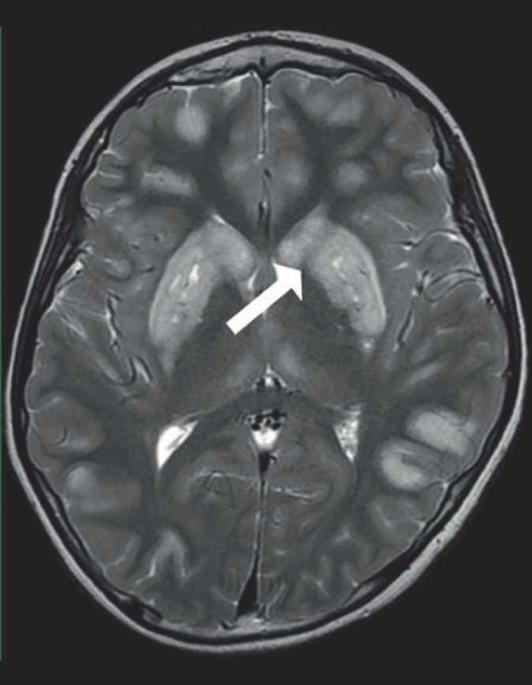
Figura 1 Imagen de RMC representativa del paciente 5. Imagen de resonancia magnética cerebral simple con corte axial con evidencia de lesiones hiperintensas a nivel de ganglios basales bilateral de forma simétrica.
La literatura reporta que en estos pacientes el pronóstico usualmente es malo, con alta mortalidad en los primeros dos años posteriores al diagnóstico, con falla ventilatoria como la principal causa, secundaria a disminución de la fuerza en músculos accesorios, lo que lleva a dificultad en el manejo de complicaciones respiratorias 1. Se han definido algunos factores de mal pronóstico entre los que se encuentran el inicio temprano en menores de 6 meses, baja talla, epilepsia farmacorresistente, niveles elevados de lactato, identificación de causa genética e ingreso a unidad de cuidados intensivos 1. Llama la atención que los pacientes analizados tienen una evolución más larga de lo usual, lo que está de acuerdo con la presentación clínica descrita para pacientes cuyo inicio de la enfermedad es posterior a los 6 meses.
Solo uno de los pacientes tiene historia familiar de casos similares y otro es producto de una pareja consanguínea. Es importante recordar que el SL tiene un origen mitocondrial, con compromiso genético heterogéneo; se han descrito mutaciones en cerca de 60 genes y aun así, hasta el momento solo se explican la mitad de los casos descritos en estos estudios 1,5,6,10,11. Estos genes afectan el sistema de fosforilación oxidativa (OXPHOS), que lleva acabo ceso de producción energética mitocondrial 1,5,6,10,11. Actualmente no se cuenta con tratamiento curativo, no existe ninguna evidencia medica científica que soporte el uso de diferentes tipos de medicamentos; sin embargo, pueden utilizarse algunas estrategias para mejorar la calidad de vida y disminuir los síntomas de la enfermedad 17.
El tratamiento con complejo B es ampliamente utilizado, ya que actúa como cofactor del complejo enzimático piruvato deshidrogenasa; el uso de biotina y tiamina ha demostrado mejoría de los síntomas en pacientes con enfermedades mitocondriales; sin embargo, en pacientes con SL la respuesta es variable y depende de la etiología genética 17. El uso de coenzima Q10 ha demostrado alguna utilidad en el manejo sintomático, aunque su respuesta varía dependiendo del genotipo 17. A partir de la coenzima Q10 se han producido algunos derivados como la idebenona y la quinona EPI-743 que presentan mejor ingreso a la mitocondria y aparentemente mejor respuesta a tratamiento, con algunos casos esporádicos de mejoría de la sintomatología 17. Los pacientes se encuentran en tratamiento con los medicamentos disponibles y, con excepción de los casos 4 y 5, se mantienen estables clínicamente. Los manejos con vitaminas del complejo B, coenzima Q10 o sus derivados pueden ayudar en algunos casos. Actualmente se han utilizado medicamentos como el benzofibrato, AICAR, resveratol y ribosa de nicotinamida 5,6 que aumentan la biogénesis mitocondrial, y han demostrado alguna utilidad cuando la cadena respiratoria no se encuentra completamente inhibida.
En resumen, dos de los pacientes presentan cuadro clínico característico de la enfermedad iniciada antes de los 12 meses de vida, y los tres pacientes restantes presentan una forma de inicio tardía que es poco frecuente en esta patología. En uno de los pacientes se presentó retraso del neurodesarrollo con posterior regresión mientras que en los restantes presentaron un adecuado desarrollo con regresión de inicio súbito. Las distonías se presentaron como síntoma preponderante; a pesar de ser un hallazgo frecuentemente reportado, solo dos presentaron epilepsia asociada al SL.
La respuesta al tratamiento en nuestros pacientes ha sido variable, sin evidenciar franca mejoría de la enfermedad como se ha demostrado en otros estudios.
Conclusión
El SL es una enfermedad neurodegenerativa catastrófica, multisistémica, con evidencia de importante heterogeneidad clínica y genética. La aproximación diagnóstica al SL, como a cualquier otra enfermedad metabólica, inicia desde la semiología clínica y se apoya en los hallazgos conseguidos a través de las diferentes herramientas disponibles bioquímica, imagenología y genética. En los casos presentados no ha sido posible realizar el análisis molecular, sin embargo, los hallazgos clínicos, bioquímicos e imagenológicos son compatibles con SL.

